Article Text

Abstract
Objectives Tolerogenic dendritic cells (tolDCs) constitute a promising experimental treatment for targeting autoreactive T cells in autoimmune diseases, including rheumatoid arthritis (RA). The authors' goal is to bring tolDC therapy for RA to the clinic. Here the authors address key translational issues related to the manufacturing of tolDCs from RA patients with current good manufacturing practice (cGMP)-compliant reagents, the stability of tolDCs, and the selection of suitable quality control markers.
Methods Human monocyte-derived tolDCs were established from RA patients and healthy controls (HCs) using the immunosuppressive drugs dexamethasone and vitamin D3, and the cGMP-grade immunomodulator, monophosphoryl lipid A, in the cGMP-compliant medium, CellGroDC. The functionality of tolDCs and tolDC-modulated autologous CD4 T cells was determined by flow cytometry, [3H]thymidine incorporation and ELISA.
Results Clinical-grade tolDCs established from patients with RA exhibit a typical tolerogenic phenotype of reduced costimulatory molecules, low production of proinflammatory cytokines and impaired stimulation of autologous antigen-specific T cells, comparable to HC tolDCs. Toll-like receptor 2 (TLR-2) was highly expressed by tolDCs but not mature DCs. Furthermore, tolDCs suppressed mature DC-induced T cell proliferation, interferon γ and interleukin 17 production, and rendered T cells hyporesponsive to further stimulation. Importantly, tolDCs were phenotypically stable in the absence of immunosuppressive drugs and were refractory to further challenge with proinflammatory mediators.
Conclusions tolDCs established from patients with RA are comparable to those derived from healthy donors. TLR-2 was identified as an ideal marker for quality control of tolDCs. Potently tolerogenic and highly stable, these tolDCs are a promising cellular therapeutic for tailored immunomodulation in the treatment of RA.
This paper is freely available online under the BMJ Journals unlocked scheme, see http://ard.bmj.com/info/unlocked.dtl
Statistics from Altmetric.com
Introduction
Rheumatoid arthritis (RA) is a chronic, debilitating autoimmune disease with no known cure. Autoimmune diseases including RA are thought to arise through a breakdown in self-tolerance. Current treatments involve non-antigen-specific global immunosuppression leading to numerous side effects. Much research has therefore focused on the development of more selective immuno-suppressive, drug-sparing therapies with fewer complications and the potential of long-term disease remission. T cell immunomodulation, or immune reprogramming, is an attractive strategy for treatment of autoimmune disorders,1 and a novel approach for targeting autoreactive T cells is the use of antigen-specific dendritic cells (DCs).2
DCs are critical in the initiation of immune responses against invading pathogens and tumours, but also in the induction of central and peripheral tolerance to self-antigens.3 The constitutive depletion of DCs in mice leads to a collapse in self-tolerance and the development of fatal autoimmune disease.4 Tolerogenic DCs (tolDCs) induce tolerance through the presentation of antigen with inadequate costimulation and cytokine production for effector T cell activation, resulting in T cell silencing or deletion or induction of regulatory T cells.5 6
tolDCs can be generated in vitro by a variety of methods, including genetic or pharmacological modification.7 Injection of ex vivo modified tolDCs has proven beneficial in models of autoimmune disease, including collagen-induced arthritis,8,–,18 diabetes,19 experimental autoimmune encephalo- myelitis16 and uveoretinitis.20 Therapy with tolDCs therefore has great potential for the treatment of autoimmunity in humans, and the current challenge is to develop tolDCs for clinical application. Others have addressed the development of clinical-grade DCs for cancer immunotherapy.21,–,23 However, criteria for the development of safe tolDCs for the treatment of autoimmune disease remain to be evaluated.
Previously, we have described a simple and robust method for the establishment of tolDCs by treatment of monocyte-derived DCs with the immunosuppressive glucocorticoid, dexa-methasone (Dex), the vitamin D receptor agonist, 1,25(OH)2D3 (vitamin D3 (VitD3)) and the toll-like receptor 4 (TLR-4) ligand, lipopolysaccharide (LPS) from Escherichia coli.24 25 These tolDCs, characterised by low expression of costimulatory molecules and low production of proinflammatory cytokines, have impaired T cell stimulatory capacity and tolerise T cells in vitro. Our aim is to conduct clinical trials with these tolDCs for RA.5 26 27
Here we address several key translational issues pertaining to clinical application of tolDCs. Firstly, research-grade reagents—for example, fetal bovine serum or LPS—need to be substituted for current good manufacturing practice (cGMP)-compatible reagents, without compromising the T cell-tolerising effects of tolDCs. Furthermore, monocyte-derived DCs are more proinflammatory in RA,28 29 therefore our protocol requires validation to guarantee generation of effective tolDCs from patients with RA. Another important consideration is the selection of reliable and easy-to-measure quality control (QC) markers. QC markers should ideally be highly expressed,30 31 exclusively by tolDCs, rather than reliant on comparative expression with mature immunogenic DCs. Finally, as tolDCs will be used to treat RA, a chronic inflammatory disease, their stability in proinflammatory environments is paramount and requires investigation. This study describes a cGMP-compatible method for generating stable tolDCs from patients with RA, resulting in a novel autologous cellular therapy for the specific modulation of autoreactive T cells for the treatment of RA.
Materials and methods
Ethics
Peripheral blood samples from healthy controls (HCs) and patients with RA were obtained with informed consent and after approval by the Newcastle and North Tyneside Research Ethics Committee 2.
Isolation of cells
Peripheral blood mononuclear cells were isolated by density centrifugation. Monocytes were positively selected from peripheral blood mononuclear cells using anti-CD14 microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany). CD4 T cells were enriched using CD4 RosetteSep (StemCell, Vancouver, BC, Canada) or by positive selection with anti-CD4 microbeads (Miltenyi Biotec).
Establishment of DCs
Monocytes were cultured at 0.5×106/ml for 7 days in cGMP-grade CellGroDC (CellGenix, Freiburg, Germany) containing penicillin and streptomycin solution (100 U/ml and 100 µg/ml, respectively) in the presence of clinical-grade granulocyte–macrophage colony-stimulating factor (50 ng/ml; Berlex, Seattle, Washington, USA) and interleukin 4 (IL-4; 50 ng/ml; Immunotools, Friesoythe, Germany). Cells were replenished on day 3 with fresh medium and cytokines, and cytokines only on day 6. To induce mature DCs (matDCs), DCs were treated with cGMP-grade monophosphoryl lipid A (MPLA) (1.0 µg/ml; Avanti Polar Lipids, Alabaster, Alabama, USA) on day 6. tolDCs were established by treatment with Dex (1 μM, Sigma Aldrich, Poole, UK) on day 3 and Dex (1 μM) and VitD3 (0.1 nM, Biomol, Exeter, UK) and MPLA on day 6. On day 7, DCs were harvested and washed extensively before functional assays were performed. DCs were loaded during the final 24 h of culture with the recall protein antigen, purified protein derivative (PPD; 1 µg/ml; Statens Serum Institut, Copenhagen, Denmark), or candidate RA autoantigen human cartilage glycoprotein 39 (HCgp39; 10 µg/ml; Organon, Welwyn Garden City, UK). Control DCs were left unloaded. For superantigen experiments, DCs were pulsed with 5 ng/ml toxic shock syndrome toxin 1 (TSST-1) for 1 h.
DC phenotyping
Cell surface expression was investigated using the following antibodies: CD14-PE (M5E2), CD40-APC (5C3), CD80-FITC (L307.4), CD86-APC (FUN-1), CD83-PE (HB15e), HLA-DR DP DQ-FITC (Tu39), all from BD Biosciences (Oxford, UK), and TLR-2-APC (TL2.1; e-Bioscience, Hatfield, UK). DCs were washed and resuspended in staining buffer (phosphate-buffered saline supplemented with 1% fetal calf serum, 2 mM EDTA and 0.01% sodium azide) in the presence of IgG (Grifols, Los Angeles, California, USA), and incubated with antibody for 30 min before acquisition on a BD FACScan and analysed using FlowJo software (Treestar).
DC cytokine production
DCs were stimulated at 1×105 DCs/ml with CD40-ligand-transfected J558L mouse cells (a gift from Professor Peter Lane, Birmingham University, UK) at a 1:1 ratio for 24 h. Cytokine production was determined in supernatants by sandwich ELISA from BD Pharmingen (Oxford, UK) (IL-6, IL-10, IL-12p70, tumour necrosis factor (TNF)α) or e-Bioscience (IL-1β, IL-23).
DC/T cell cultures
DC/T cell cultures were carried out in complete medium: RPMI-1640 containing human AB+ serum (5%), L-glutamine (2 mM), and penicillin (100 U/ml)and streptomycin (100 µg/ml). T cells (105) were cultured with unloaded or antigen-loaded tolDCs or matDCs. In restimulation experiments, primed T cells were subsequently rested from day 6 to 10 in the presence of 20 IU/ml recombinant IL-2 (Novartis, Camberley, UK) and restimulated on day 10 with antigen-loaded autologous matDCs for 3 days. DC/T cell cultures were performed at varying ratios. Proliferation was determined by incorporation of [3H]thymidine for 18 h or by carboxyfluorescein succinimidyl ester (0.5 µM) dilution analysis of T cell receptor β-chain variable region 2 (Vβ2)-expressing cells (anti-TCR-Vβ2; clone MPB2D5; Immunotech, Marseille, France) at various time points. For the tolDC-suppression assay, additional tolDCs or matDCs were incorporated as indicated. Cytokine production in T cell supernatants was determined by sandwich ELISA for interferon γ (IFNγ) (BD Pharmingen) and IL-17 (e-Bioscience).
Intracellular cytokine staining
T cells restimulated with matDCs for 3 days were activated for maximal cytokine production with phorbol myristate acetate (50 ng/ml) and ionomycin (1 µg/ml) for 5 h in the presence of brefeldin A (10 µg/ml) for the final 4 h. Cells were stained for cell surface Vβ2 expression before fixation and permeabilisation (FoxP3 staining kit; e-Bioscience) and subsequent staining for intracellular IL-17 (clone eBio64DEC17) and IFNγ (clone B27). Cells were acquired on a BD LSRII and analysed using FlowJo software.
DC stability
Phenotypic stability of DCs was investigated 24 h after washing and reculture of DCs in complete medium. Firstly, 1×106 cells/ml were left unstimulated or stimulated with a cocktail of proinflammatory cytokines containing 1000 U/ml IFNγ and 10 ng/ml each of IL-1β, IL-6 and TNFα, 0.1 µg/ml LPS or 10 µg/ml peptidoglycan (PGN). Cells were stained as described for DC phenotyping, and cytokine production was investigated in the absence and presence of proinflammatory cytokines, LPS or PGN as described for DC cytokine production.
Statistical analysis
The following statistical analyses were performed using Prism 4.0: analysis of variance (ANOVA) for comparisons between multiple groups, Mann–Whitney U or Student t test for comparisons between two groups, and two-way ANOVA for multiple groups over time.
Results
cGMP-tolDCs from patients with RA are phenotypically and functionally comparable to HC tolDCs
Our initial studies investigated the surface phenotype, cytokine profile and T cell-stimulatory capacity of tolDCs from RA patients as compared with HCs, established using the cGMP-medium CellGroDC and bacterial immunomodulator MPLA. Immature DCs (iDCs; figure 1A) expressed reduced levels of major histocompatibility complex II (MHC II) and low levels of CD40, CD83 and the costimulatory molecules CD80 and CD86. Expression of all these markers was substantially enhanced upon DC maturation (matDCs; figure 1A,B). In contrast, tolDCs from both RA patients and HCs displayed a typical semimature phenotype of reduced MHC II and CD80 and significantly lower CD40, CD83 and CD86 expression than matDCs (figure 1B). A highly expressed marker on tolDCs was TLR-2, which was low on iDCs and matDCs (figure 1A,B) and may therefore be suitable for QC. Despite the high propensity of RA-matDCs to produce TNFα, production of all other cytokines investigated was comparable between RA and HC cultures (figure 2A). tolDCs from both RA patients and HCs consistently produced lower levels of proinflammatory cytokines IL-1β, IL-6, IL-23 and TNFα upon CD40-mediated activation, as compared with matDCs, while IL-12 was notably undetected in all tolDC cultures (figure 2A). Similar differences were found when cytokine production by tolDCs and matDCs was measured directly after maturation with MPLA, before activation through CD40 (online supplemental figure 1). Anti-inflammatory IL-10 production by tolDCs was non-significantly reduced as compared with their equivalent matDC counterparts.

Rheumatoid arthritis (RA)-derived tolerogenic dendritic cells (tolDCs) exhibit a semimature phenotype and are comparable to healthy control (HC) tolDCs. (A) HC- and RA-derived DC expression of maturation-associated markers of immature DCs (iDCs), mature DCs (matDCs) and tolDCs. Histograms are representative of five independent donors. (B) Mean expression of HC and RA patient tolDCs and matDCs is shown relative to iDCs. Expression of each marker (geometric mean fluorescence intensity (GMFI)) was normalised relative to expression by iDCs (100%) for each independent donor. Phenotypic analysis of maturation-associated markers was determined 24 h after maturation with 1 µg/ml monophosphoryl lipid A in the presence or absence of dexamethasone and vitamin D3. Data are expressed as the mean±SEM for five independent donors. Mean GMFI for HC iDCs vs RA iDCs are: MHC II, 949.8±161.3 vs 1110±178.3; CD40, 19.8±7.4 vs 20.4±6.6; CD80, 12.3±1.7 vs 11.2±1.4; CD83, 7.9±0.1 vs 9.4±1.2; CD86, 11.5±2.5 vs 21.6±5.8; toll-like receptor 2 (TLR-2), 89.9±11.1 vs 76.2±4.2. No significant difference was detected between RA and HC DC populations. *p≤0.05, **p≤0.01, ***p≤0.001 (analysis of variance). MHC, major histocompatibility complex; TLR-2, toll-like receptor-2.

Healthy control (HC) and rheumatoid arthritis (RA) tolerogenic dendritic cells (tolDCs) have an anti-inflammatory phenotpye. (A) Mean cytokine production by tolDCs and mature DCs (matDCs) in response to CD40 activation. At 24 h after maturation with 1 µg/ml monophosphoryl lipid A (MPLA) in the presence or absence of dexamethasone (Dex) and vitamin D3 (VitD3), DCs were washed, and 105 tolDCs or matDCs cocultured at a 1:1 ratio with the J588L-CD40L-expressing cell line in the absence of Dex and VitD3 for a further 24 h. Supernatants were harvested for cytokine analysis by ELISA. Data are expressed as the mean±SEM for at least four HCs and seven RA-independent donors. (B) Representative RA donor DC-derived cytokine production when unloaded (UL) or loaded with 10 µg/ml human cartilage glycoprotein 39 (HCgp39) as a model antigen, during maturation with 1 µg/ml MPLA. Unloaded and antigen-loaded DCs were activated via CD40 as described in (A). Data are representative of four independent donors. Cytokine production was determined by ELISA. <d, below limits of detection. Detection limit for IL-12 is 30 pg/ml. *p≤0.05, **p≤0.01, ***p≤0.001 (Mann–Whitney U test). TNF, tumour necrosis factor.
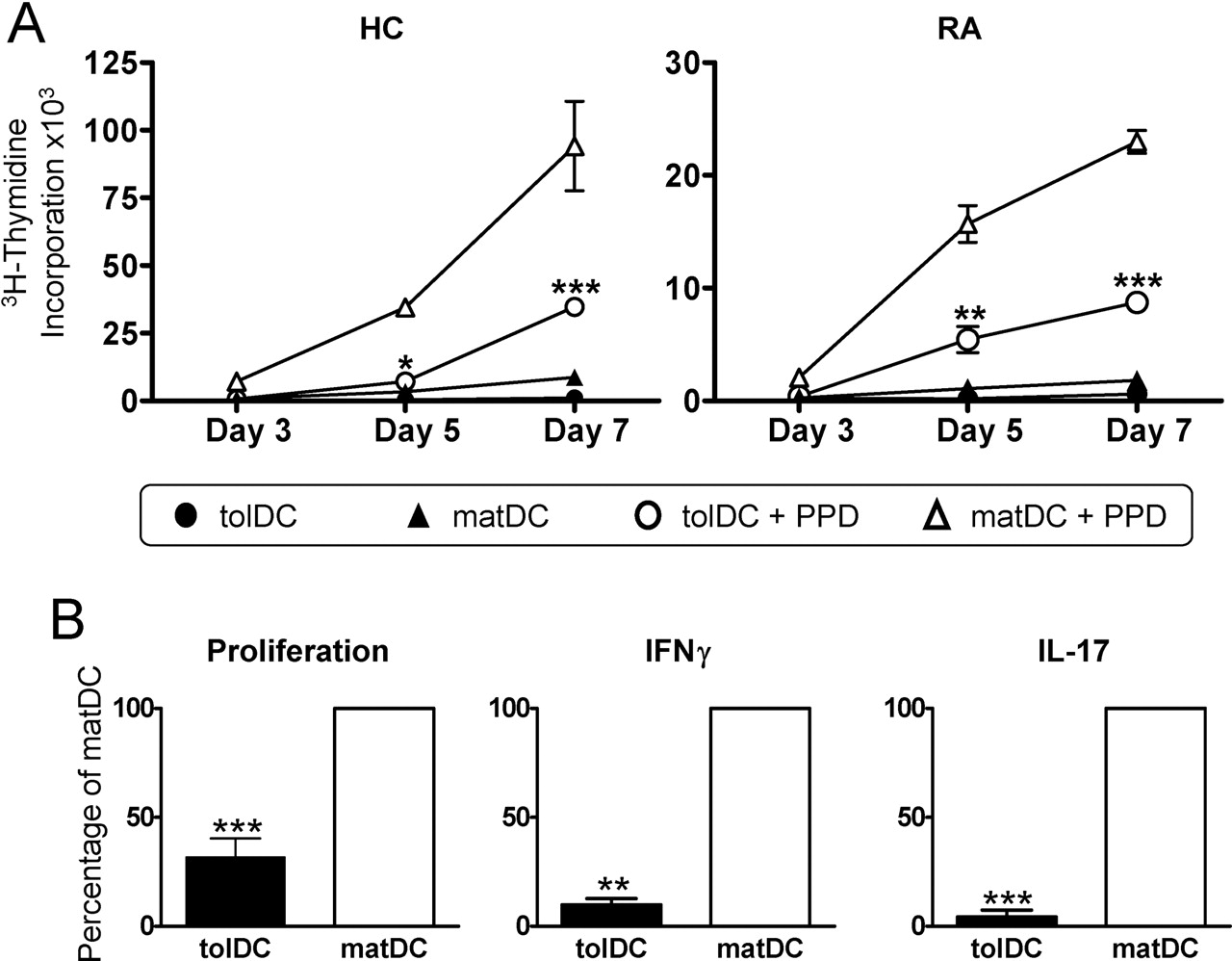
The cytokine profile of tolDCs was unaffected by incorporation of an antigen-loading step during tolDC generation, investigated using a RA candidate autoantigen (HCgp39; figure 2B). Furthermore, we investigated the autologous T cell immunostimulatory capacity of tolDCs using the recall antigen, PPD. Minimal T cell proliferation was observed in response to both RA and HC non-antigen-loaded DC populations, while PPD-loaded matDCs induced significant T cell proliferation. Consistent with their semimature phenotype and anti-inflammatory cytokine profile, both RA and HC PPD-loaded tolDCs only poorly induced antigen-specific proliferation (figure 3A,B) and IFNγ and IL-17 production by autologous T cells (figure 3B).

Antigen-specific T cells exhibit a reduced responsiveness to tolerogenic dendritic cells (tolDCs). (A) 105 healthy control (HC) and RA CD4 T cells were stimulated with 104 autologous unloaded or purified protein derivative (PPD)-loaded (1 µg/ml) tolDCs or mature DCs (matDCs), and proliferation determined at various time points. Data are expressed as the mean±SEM of triplicate wells, representative of three independent experiments. (B) Mean healthy control (HC) CD4 T cell proliferation and cytokine production as a percentage of matDC-induced T cell responses. HC CD4 T cells (105) were stimulated at a 10:1 ratio with 104 PPD-loaded tolDCs or matDCs. Cytokine production and proliferation were determined on day 6 and 7, respectively. Data are expressed as the mean of three independent experiments. Mean values for matDC-induced proliferation were 33 642±18807 cpm, interferon (IFN)γ was 16.9±8.1 ng/ml and interleukin (IL)-17 was 359.5±328.8 pg/ml. T cell proliferation was determined by [3H]thymidine incorporation, and cytokine production was quantitated by ELISA. *p≤0.05, **p≤0.01, ***p≤0.001 as compared with matDCs (two-way analysis of variance (A) and Student t test (B)).
tolDCs induce T cell hyporesponsiveness
We further investigated the immunoregulatory capacity of clinical-grade tolDCs in a superantigen-based T cell activation model. This model is well established for studying T cell activation and modulation by autologous DCs.32,–,36 TSST-1 binds with high affinity to TCR-Vβ2, preferentially expanding Vβ2+ T cells in a MHC II-dependent manner,37 although expansion of low-affinity Vβ2− T cells can occur.32 Here we quantitated the T cell response to tolDCs and matDCs loaded with TSST-1 (figure 4A). matDCs were superior over tolDCs in inducing T cell proliferation, inducing activation of Vβ2+ T cells even at very low DC numbers and also inducing expansion of Vβ2− T cells at higher DC numbers. In contrast, tolDCs expanded Vβ2+ T cells at high DC concentrations, but failed to support strong Vβ2− T cell proliferation (figure 4A). Upon optimal restimulation with TSST-1-loaded matDCs, T cells primed by TSST-1-loaded tolDCs (Ttol) showed impaired proliferative potential (figure 4B) and a reduction in the percentage of IFNγ- and IL-17-producing T cells as compared with T cells primed by TSST-1-loaded matDCs (Tmat) (figure 4C), suggestive of Ttol hyporesponsiveness.

Tolerogenic dendritic cells (tolDCs) induce T cell hyporesponsiveness. (A) 105 CD4 T cells were primed with the indicated numbers of toxic shock syndrome toxin 1 (TSST-1)-loaded (5 ng/ml) autologous tolDCs or mature DCs (matDCs) and proliferation of carboxyfluorescein succinimidyl ester (CFSE+) T cell receptor β-chain variable region 2 (TCR-Vβ2)-stained cells quantitated at day 5. Values represent percentage of cells as a percentage of CD4 T cell population. Percentage of Vβ2+ cells in starting CD4 population was 8.9±0.95%. (B) and (C) T cells primed with TSST-1-loaded (5 ng/ml) tolDCs (Ttol) or matDCs (Tmat) (10:1 ratio) for 6 days were rested with interleukin (IL)-2 (20 IU/ml) for 4 days before CFSE labelling and restimulation at a 10:1 ratio with TSST-1-loaded matDCs on day 10. (B) Day 13 T cell proliferation was determined by CFSE dilution. Values represent the percentage of proliferating cells. (C) Day 13 intracellular IL-17 and interferon (IFN)γ production was quantitated after 5 h stimulation with phorbol myristate acetate (PMA) (50 ng/ml) and ionomycin (1 µg/ml) with brefeldin A (10 µg/ml) added for the final 4 h. Values represent the percentage of cytokine-positive cells as a percentage of the total CD4 T cell population (Ttot). Gates determining positive cells were set using equivalent non-PMA/ionomycin-stimulated controls. The percentage of positive cells for IL-17 and IFNγ unstimulated cultures was <1% and <2%, respectively. Data shown are representative of two independent experiments.
tolDCs suppress matDC-induced T cell proliferation, IFNγ and IL-17
Successful tolDC therapy may in part depend on the ability to suppress immune responses induced by resident immunogenic DCs. We therefore investigated the ability of tolDCs to modulate matDC-induced PPD-specific T cell proliferation and cytokine production. Co-incubation of PPD-loaded tolDCs and matDCs significantly inhibited T cell proliferation (figure 5A), IFNγ and IL-17 production (figure 5B) at a 1:1 tolDC/matDC ratio and significantly inhibited proliferation (figure 5A) and IFNγ production at a 1:10 tolDC/matDC ratio (figure 5B) as compared with PPD-loaded matDCs alone. These inhibitory effects were not caused by increasing the total DC number, as doubling the number of matDCs did not result in suppression of T cell proliferation and even enhanced cytokine production (figure 5A,B).
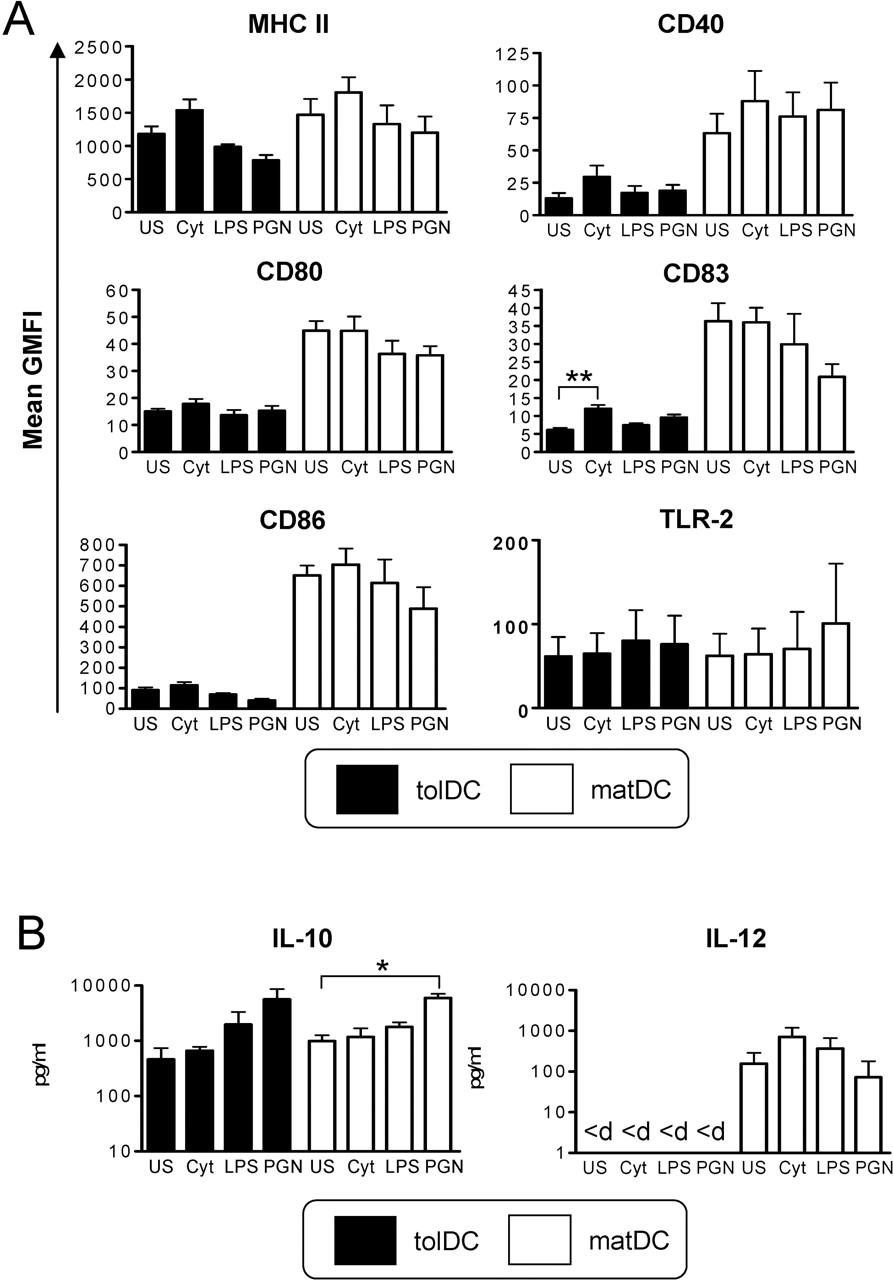
Tolerogenic dendritic cells (tolDCs) suppress mature DC (matDC)-induced T cell proliferation, interferon (IFN)γ and interleukin (IL)-17. (A) Representative graph showing control CD4 T cell responses to purified protein derivative (PPD)-loaded DCs (1 µg/ml). CD4 T cells (105) were cocultured at a 10:1 ratio with tolDCs or matDCs alone or matDCs with an equivalent number of tolDCs or matDCs as a cell number control (1:1 ratio; 104) or 10-fold fewer tolDCs than matDCs (1:10 ratio; 103). Proliferation was quantitated at days 3, 5 and 7 by [3H]thymidine incorporation. The graph is representative of four independent experiments. (B) Mean IFNγ and IL-17 cytokine production relative to matDCs. CD4 T cells (105) were stimulated with autologous PPD-loaded DCs as in (A). Cytokine production was quantitated on day 6 by ELISA. Data are expressed as the mean±SEM from three independent experiments. *p≤0.05, **p≤0.01, ***p≤0.001 as compared with matDCs (two-way analysis of variance (A) and analysis of variance (B)).
tolDCs have a highly stable semimature, anti-inflammatory phenotype
A key consideration in the development of a tolDC therapy for RA and other chronic inflammatory diseases is the stability of tolDCs to proinflammatory mediators. tolDC stability was therefore investigated after removal of Dex, VitD3 and MPLA and reculture in medium containing human serum. Both tolDCs and matDCs were phenotypically refractory to stimulation with all proinflammatory mediators investigated, with tolDCs retaining their typical semimature phenotype and cytokine profile (figure 6A,B). TLR-2 expression by tolDCs was found to be downregulated comparable to that of matDC expression (figure 6A), while the only marker that was significantly upregulated by tolDCs was CD83 in response to proinflammatory cytokines, although levels remained significantly (p<0.01; paired t test) lower than that expressed by equivalent matDCs (figure 6A). CD40-activation-induced cytokine production by tolDCs and matDCs was unaltered in response to cytokine or LPS challenge (figure 6B), while PGN induced increases in IL-10 production by both tolDCs and matDCs. IL-12p70 remained undetectable in tolDC cultures under all conditions investigated (figure 6B). Overall, these data indicate that tolDCs are highly stable.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Tolerogenic dendritic cells (tolDCs) have a highly stable semimature, anti-inflammatory phenotype. Phenotypic stability of tolDCs and mature DCs (matDCs) was determined in response to inflammatory stimuli. tolDCs or matDCs were washed and recultured in the absence of tolerising factors including dexamethasone and vitamin D3 for 24 h. Cultures were either unstimulated (US) or stimulated with a cocktail of proinflammatory cytokines (cyt) containing interleukin (IL)-1β, IL-6, tumour necrosis factor (TNF)α (all 10 ng/ml) and interferon (IFN)γ (1000 U/ml), lipopolysaccharide (LPS) (0.1 µg/ml) or peptidoglycan (PGN) (10 µg/ml). (A) DCs were analysed for cell surface phenotype by flow cytometry. Plots show mean geometric mean fluorescence intensity (GMFI) of DC-maturation markers of tolDCs and matDCs. Data are expressed as the mean GMFI±SEM of at least four independent donors. (B) tolDCs and matDCs (105/well) were activated via CD40 by culture with an equivalent number of the J588L-CD40L-expressing cell line in the presence or absence of inflammatory mediators and supernatants collected after 24 h for analysis by ELISA. <d, below limits of detection (30 pg/ml for IL-12 ELISA) *p<0.05, **p≤0.01 (analysis of variance). MHC, major histocompatibility complex; TLR-2, toll-like receptor 2.
Discussion
This study describes the development of clinical-grade tolDCs, focusing on a number of key translational issues that require elucidation before therapeutic application of these cells.
We investigated the functionality of tolDCs generated using cGMP-grade reagents, suitable for the development of an Investigational Medical Product for human therapy. We describe a protocol for the establishment of semimature, anti-inflammatory, clinical-grade tolDCs using the cGMP-compliant serum-free medium CellGroDC and DC immunomodulator, MPLA. These clinical-grade tolDCs are comparable to our previously characterised research-grade tolDCs24 25 in terms of cell surface phenotype, cytokine production and in vitro T cell modulatory function. CellGroDC was deemed the medium of choice23 and is currently used in DC vaccine development for cancer.38,–,40 MPLA, a low-toxicity alternative to LPS, is currently being tested in vaccine development.41 As a known TLR-4 agonist inducing matDCs,42 MPLA is less potent than LPS,43 which may explain the increased MPLA dose (1.0 µg/ml MPLA vs 0.1 µg/ml LPS25) required to induce comparable DC maturation.42,–,44 We observed that MPLA potently induces IL-10 production by DCs. Considering the inhibitory effect of IL-10 on IL-12p35 and p40 mRNA45 46 and IL-12p40 protein,46 it is not surprising that MPLA only poorly induced IL-12p70 and IL-23 (consisting of IL-12p40 and IL-23p19) by matDCs in this study. As the lack of IL-12 production by tolDCs is pivotal to their tolerogenicity,24 47 MPLA by virtue of its inability to induce IL-12 is highly appropriate for the establishment of tolDCs, but less suitable for the generation of matDC therapy for cancer.
As antigen-presenting cell dysregulation has been implicated in RA pathogenesis,48 we confirmed that this protocol could be used for the establishment of tolDCs from patients with RA. Indeed, in our and other studies,28 29 matDCs established from patients with active RA produced significantly more TNFα than HCs or RA patients in remission. We demonstrate that, despite an inherently more proinflammatory cytokine profile, tolDCs established from RA patients adopt a HC-tolDC-like phenotype, suggesting that, even with potentially altered antigen-presenting cell functionality during RA, it remains possible to establish tolDCs ex vivo for autologous cellular immunotherapy. To our knowledge, this is the first study to demonstrate the generation of tolDCs from RA patients.
tolDCs for immunotherapy must be safe, standardised and controlled30 31; however, robust and reliable markers are poorly defined. Ideally, QC of tolDCs should be based on markers that are quickly and readily detectable. Phenotypic identification of tolDCs is most commonly determined relative to other DC populations (eg, iDCs and/or matDCs47), which is impractical in the cGMP laboratory setting. This study revealed the exclusive high-level expression of TLR-2 by tolDCs, a known response to glucocorticoids,49,–,51 making this an ideal marker for QC purposes.
Of particular importance in tolDC treatment of inflammatory autoimmune diseases including RA is their stability. We therefore investigated the stability of tolDCs to a number of known proinflammatory mediators in vitro, as the use of maturation-sensitive tolDCs in ongoing inflammatory disease risks the conversion of tolDCs into matDCs with the potential to exacerbate disease.52 This study revealed that cGMP-tolDCs, despite differential signalling of MPLA and LPS via TLR-4,43 were rendered refractory to further maturation with LPS, a phenomenon previously described for LPS as LPS desensitisation.53 tolDCs were also resistant to maturation and maintained stable cytokine production in response to stimulation with proinflammatory cytokines, a process termed DC exhaustion or paralysis.33 54 55 Even though the tolDCs described here express high levels of TLR-2, PGN, a microbial compound recognised by TLR-2,56 had only subtle effects on tolDCs. Consistent with our findings, it was recently shown that LPS-activated, Dex-treated DCs further challenged with LPS in the absence of Dex were phenotypically stable and remained poor producers of TNFα and IL-6.57 However, other TLR ligands or proinflammatory cytokines were not investigated in that study. In contrast with the study of Chamorro et al,51 our study revealed a downregulation of TLR-2 expression in the absence of Dex, and TLR-2- or TLR-4-mediated challenge of tolDCs failed to enhance TLR-2 expression by tolDCs. This may be explained by the different TLR-2 ligands used or be due to the different protocols used for the establishment of tolDCs. Importantly, the cGMP-tolDCs described here, once removed from the tolerising environment of Dex, VitD3 and MPLA, maintained a typical semimature phenotype and failed to secrete detectable IL-12 in response to a variety of DC maturation factors, making them well suited for the treatment of ongoing inflammatory disease.
A major advantage of tolDC immunotherapy is the potential for antigen-specific T cell tolerisation, thereby minimising the need for sustained non-specific therapies with side effects. Our study demonstrates that tolDCs suppress immunogenic DC-induced T cell proliferation at a 1:10 tolDC/matDC ratio in vitro. Clinical trial of tolDC therapy will ultimately reveal tolDC efficacy; however, localised administration of tolDCs to the arthritic joint will facilitate high tolDC/matDC ratios, which may be required for clinical efficacy and the re-induction of T cell tolerance. Using model antigens, we demonstrate that tolDCs are unaffected by antigen loading and mediate antigen-specific hyporesponsiveness without inducing proliferation of ‘bystander’ T cells with low affinity for the antigen, all of which are important features of targeted antigen-specific therapy. The pathogenic autoantigen in RA is unknown, however; future studies will reveal the tolerising effects of tolDCs to non-characterised RA autoantigen(s) present within synovial fluid.58,–,60
This study describes the development of clinical-grade tolDCs established using a simple and robust protocol. These tolDCs are classically semimature with an anti-inflammatory phenotype and exhibit high-level expression of TLR-2, highly appropriate for QC. Potently tolerogenic to autologous T cells, these cGMP-tolDCs are highly stable in proinflammatory environments. tolDCs are a promising novel autologous cellular therapy for the specific modulation of autoreactive T cells in the treatment of RA and other inflammatory autoimmune diseases.
Acknowledgments
We thank Mr T Wooldridge and Dr G Raftery for the recruitment of patients with RA, and J Diboll for technical assistance. Mr T Wooldridge is funded by the UK NIHR Biomedical Research Centre for Ageing and Age-related disease award to the Newcastle upon Tyne Hospitals NHS Foundation Trust. This work was supported by the JGW Patterson Foundation and Arthritis Research UK (grant 17750).
References
Footnotes
-
Funding JGW Patterson Foundation, Arthritis Research Campaign.
-
Competing interests None.
-
Ethics approval This study was conducted with the approval of the Newcastle and North Tyneside Research Ethics Committee 2.
-
Provenance and peer review Not commissioned; externally peer reviewed.