Article Text

Abstract
Background: Synovial tissues in patients with Chlamydia associated arthritis are persistently infected by C trachomatis, an organism for which genetic manipulation is not possible. M tuberculosis also engages in persistent infection, and because this bacterium is genetically tractable many groups have been able to define transcriptional characteristics of mycobacterial growth and persistence.
Objective: To investigate whether the pattern of gene expression underlying chlamydial persistence is similar to that underlying mycobacterial persistence.
Methods: 194 genes in M tuberculosis that are transcriptionally up regulated to support in vivo growth and persistence of that organism have previously been identified. Each of those genes was compared with the C trachomatis genome to identify orthologues. Expression of selected chlamydial orthologues so identified was assessed by real time RT-PCR in an in vitro model of chlamydial persistence and synovial tissues from patients who were PCR positive for C trachomatis at that site.
Results: 67 C trachomatis genes were identified as being orthologous to mycobacterial persistence related genes, representing 35% of the genes tested. The chlamydial orthologues fell into similar metabolic and other categories as those in M tuberculosis. Expression of a majority of selected chlamydial orthologues was strongly up regulated in an in vitro model of chlamydial persistence and in synovial tissues of relevant patients, compared with their expression during active infection.
Conclusions: These observations provide new insight into the molecular genetic basis underlying chlamydial persistence, and indicate that this information can be obtained, in some instances, by extrapolating observations made in other biological systems and/or organisms.
- pathogenesis
- persistent infection
- gene expression
- reactive arthritis
- Chlamydia trachomatis
Statistics from Altmetric.com
An obligate intracellular pathogen, Chlamydia trachomatis, is the most prevalent sexually transmitted bacterium in America and other developed countries.1 An often severe acute inflammatory arthritis develops in some subjects with a prior urogenital infection with this organism, and about half of all patients who develop acute arthritis progress to chronic disease (reviewed by Villareal et al2 and Gérard et al3). The reason(s) that only a limited number of patients with a genital chlamydial infection develop acute arthritis is/are poorly understood. We also do not understand why only a portion of subjects with acute disease progress to chronicity, although both issues relate to our lack of detailed understanding of host-pathogen interaction during synovial chlamydial infection.
C trachomatis undergoes a biphasic developmental cycle at its sites of primary infection, the urethral or cervical epithelium (reviewed by Hatch4). This organism can disseminate widely from those primary infection sites, and when it does so the vehicle of its dissemination is the monocytic cell, the cell type which is the primary synovial host in both acute and chronic Chlamydia-induced arthritis.4,5 In contrast with the normal progression through the developmental cycle undergone during active infection of epithelial cells, however, C trachomatis residing within monocytic cells in the joint pass rapidly into an unusual biological state designated “persistence”.3 In this state, the organisms are morphologically aberrant, non-culturable by standard laboratory methods, resistant to antibiotics, and they display an unusual transcript profile.6,7,8,9,10,11,12C pneumoniae, a human respiratory pathogen related to C trachomatis, has also been implicated in eliciting inflammatory arthritis, and this organism too has been shown to cause persistent infection under some circumstances.13,14
It seems clear that synovial pathogenesis in Chlamydia-induced arthritis is a function of persistent, rather than normal active, infection. However, details relating to the mechanism(s) by which chlamydiae enter, and thereafter maintain, the persistent infection state in vivo, as well as details about the means by which persistent chlamydiae elicit disease, remain to be elucidated. As mentioned, persistent chlamydiae display an unusual transcript profile compared with that of active infection. For example, expression of omp1, the gene encoding the major outer membrane protein, is severely attenuated in persistence.7 Genes encoding products required for DNA replication (for example, dnaA, polA, and others) are transcribed in persistently infecting C trachomatis cells as they are during active infection, but expression of genes specifying products required for cytokinesis is severely down regulated during persistence (for example, ftsK, ftsW8). Several other chlamydial genes of known function are also differentially expressed in persistent v active infection.3,9,10 Full understanding of the genetic programme underlying entry into and maintenance of chlamydial persistence will be a difficult task, because many coding sequences on the C trachomatis and C pneumoniae genomes specify products of unknown function.15,16 To complicate matters further, no system for genetic manipulation of either pathogen exists to date.
Bacterial pathogens other than C trachomatis and C pneumoniae take part in persistent infection, and in some cases the mechanisms by which those organisms establish persistence have been studied extensively. Mycobacterium tuberculosis, in which genetic manipulation is possible and for which the full genome sequence is available, is one such organism.17,18. Studies from several laboratories have defined many critical aspects underlying mycobacterial persistence.19–21 For example, one recent investigation of genes required for in vivo growth and persistence of M tuberculosis in an animal model of disease identified 194 coding sequences required for those processes.22 Virtually all of the genes identified in that study as being required for establishment of persistence were not expressed, or were expressed at only low level, during axenic growth of the organism.
Because genetic manipulation of Chlamydia is not available, identification of chlamydial genes orthologous to those shown to function in persistence in other organisms may be of value. We undertook a study to determine whether none, some, or all of the 194 genes shown to be transcriptionally up regulated in support of growth and persistence in vivo for M tuberculosis have orthologues in C trachomatis. We report that 35% of the coding sequences identified in the mycobacterial study have related genes in C trachomatis. We further show that a selected set of those chlamydial orthologues spanning several metabolic and other categories show up regulation of expression during persistence, as in M tuberculosis, in both an in vitro model of that infection state and in samples from patients with arthritis who are polymerase chain reaction (PCR) positive for the organism in synovial tissue.
PATIENTS AND METHODS
Patient samples
Synovial biopsy specimens were procured under an approved protocol from patients attending the Arthritis Clinics at the V.A. Medical Center and the University of Pennsylvania Hospital, Philadelphia PA, USA; protocol approval was from the University of Pennsylvania School of Medicine and Wayne State University School of Medicine IRB committees. Synovial biopsy specimens were obtained by the Parker-Pearson method and were immediately snap frozen at −80°C until prepared for analyses.23 The three patients studied were included solely because they were PCR positive for C trachomatis chromosomal DNA in synovial tissues, and because enough tissue was present from each to support the several molecular genetic analyses required. Diagnoses for the patients were made according to criteria of the American College of Rheumatology. Table 1 summarises the clinical characteristics of each patient from whom samples were obtained.
Characteristics of patients included in this study
Cell growth, in vitro infection with C trachomatis
HEp-2 cells support active growth of C trachomatis8,14—that is, Chlamydia infecting this cell line progress normally through the developmental cycle. Nearly confluent monolayers of HEp-2 cells were infected with C trachomatis K serovar at a multiplicity of infection of 1:1 in the standard manner.8,9 Infected cultures were harvested at 12 hours after infection, and cell pellets were snap frozen at −80°C until processed for analysis. C trachomatis infecting normal human peripheral monocytes in culture enter the persistent infection state by day 3 after infection.7,8,9,10 Blood samples were procured from volunteer donors under an approved protocol, and monocytic cells were prepared and entered into culture as described by us6,7,8,9,10; the protocol was approved by the Wayne State University School of Medicine IRB. Nearly confluent monolayers of monocytes were infected at a multiplicity of infection of 1:1 with C trachomatis serovar K, and infected cultures were harvested at day 5 after infection; cell pellets were snap frozen at −80°C until processed. To be certain that chlamydiae infecting monocytes in these experiments had entered the persistent state, transcripts from the C trachomatisdnaA, polA, omp1, ftsK, pyk, and gap genes were assessed by reverse transcriptase (RT)-PCR (see just below); all such transcripts were absent as expected, except those from dnaA and polA, which are expressed during both active and persistent infection.8
Preparation and analysis of RNA/cDNA
Total nucleic acid preparations were made from Chlamydia infected HEp-2 and human monocyte cell pellets, and from synovial tissue samples, as described previously.8,9 Pure RNA was prepared from aliquots of each preparation by treatment with DNaseI (RQ1 DNase; Promega Biotech, Madison WI, USA), followed by extraction in phenol:chloroform, and collection by ethanol precipitation. Reverse transcription of total RNA preparations to cDNA was done using the MuLV enzyme and random hexamers as primers (Invitrogen, Carlsbad CA, USA), as described.10,14
cDNA preparations were subjected to quantitative real time RT-PCR analyses for 16 targeted C trachomatis genes, using the SYBR green method.10,14 Table 2 shows the coding sequences of interest and the primer systems used in the analyses. Each real time RT-PCR assay for each targeted gene was run in triplicate independently. Data were normalised to chlamydial 16S rRNA, as described8,10,14; data for specific transcripts in all monocyte and patient derived assays were indexed to the level of that transcript in C trachomatis actively growing for 12 hours in HEp-2 cells. Assays were done using a PE Biosystems (Foster City, CA, USA) model 7700 sequence detector; data were analysed using version 1.9 sequence analysis software from PE Biosystems.
C trachomatis genes targeted and primer sequences for real time RT-PCR
RESULTS
C trachomatis genes orthologous to M tuberculosis persistence related genes
To determine whether any of the 194 genes from M tuberculosis identified as being required for growth and persistence in vivo in mice22 have orthologues in C trachomatis, we performed a BLAST evaluation of each identified mycobacterial coding sequence against the full chlamydial genome (http://www.stdgen.lanl.gov, accessed 7 December 2005). Of the 194 genes, we identified 67 orthologues with Expect values of 10−1 or better, representing 35% of the total number of genes assessed. Table 3 provides a complete list of the chlamydial coding sequences identified, their related genes in M tuberculosis, and the Expect values. The chlamydial orthologues of mycobacterial persistence related genes identified fall into general categories similar to those in the M tuberculosis study—that is, genes encoding products involved in cell envelope synthesis or modification, synthesis of cofactors, transport, translation, and so on.22 Interestingly, nine genes specifying proteins of unknown function were identified on the C trachomatis genome, all but one of which corresponded to similarly unknown genes in M tuberculosis. No genes specifying components of the chlamydial type III secretion system were identified, but secA, a component of an apparent type II secretion system, was identified (but see below). Three secretion related mycobacterial genes were shown to be up regulated in the earlier study, including secA2.
C trachomatis genes identified in the BLAST search using M tuberculosis persistence related genes as query
A few potentially important differences emerged in the panel of chlamydial orthologues identified. For example, we showed earlier that expression of genes encoding products for the glycolytic and pentose phosphate pathways is severely attenuated in persistent C trachomatis cells,9 while transcription of several genes involved in energy production and conversion were shown to be up regulated in the mycobacterial study. Importantly, 107 of the 194 genes identified in the mycobacterial study as required for in vivo growth and persistence were coding sequences of unknown function; the majority of these 107 genes had no orthologous coding sequence in C trachomatis (see “Discussion”).
Expression of selected C trachomatis orthologues during persistent infection in vitro
To confirm that the Chlamydia genes identified are not only expressed during persistent infection but also up regulated over their level of expression during active infection, we selected 16 chlamydial orthologues from various functional categories and assessed their relative expression level by real time RT-PCR during both infection states; the genes subjected to this analysis are indicated in bold in table 3.
In previous studies of chlamydial persistence, we have employed an in vitro model system using normal human monocytic cells infected with C trachomatis serovar K; in this system, chlamydiae transit from a more or less normal infection state to the persistent state by 3 days after infection.7,8,9,10 We analysed RNA from infected monocytes at day 5 after infection and compared transcript levels from each of the 16 targeted genes with the level of their expression at 12 hours after infection during active infection of HEp-2 cells. Previous microarray based studies have shown that all coding sequences on the C trachomatis chromosome and plasmid are expressed at some time during the normal developmental cycle.24,25 Consistent with those data, each of the 16 genes assessed here was being transcribed at 12 hours after infection in HEp-2 cells (data not shown).
Figure 1 provides a summary of the transcript level at day 5 after infection in the in vitro monocyte model system for each of the 16 chlamydial genes targeted relative to their expression level during active HEp-2 cell infection. Interestingly, all but four of the chlamydial genes assayed showed relatively strong up regulation during persistence in comparison with expression during active infection. The four genes showing no/minimal transcriptional up regulation during persistence were Ct762 (murC/murF, an apparent fusion protein with MUR-NAc-l-alanine and d-alanine-d-alanine ligase activity), Ct701 (secA, preprotein translocase subunit A protein), Ct624 (mviN, a putative virulence factor), and Ct868 (hypothetical protein, possibly a membrane thiol protease). Four other chlamydial orthologue genes showed powerful up regulation of expression during persistence, consistent with data from the mycobacterial study. These were Ct820 (ftsY, component of signal recognition particle), Ct727 (zntA/cadA, cation-transporting ATPase), Ct393 (proS, prolyl-tRNA synthetase), and Ct052 (hemN, oxygen independent coproporphyrinogen III oxidase). All other chlamydial genes assessed showed moderate to fairly strong transcriptional up regulation in persistent C trachomatis compared with that shown in actively infecting organisms.
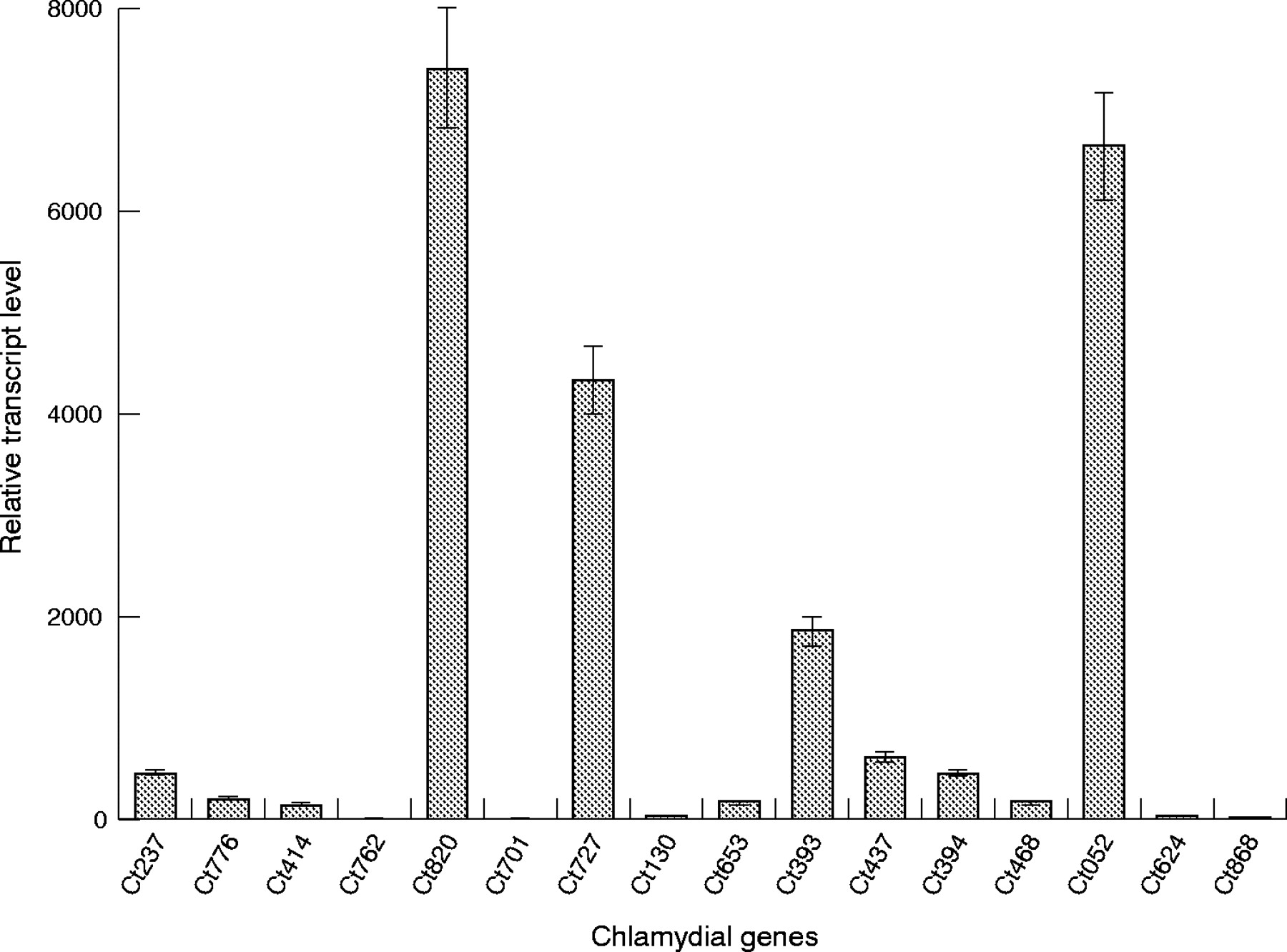
Real time RT-PCR analysis of transcript levels from 16 C trachomatis genes identified as orthologous to M tuberculosis genes required for in vivo growth and persistence, using cDNA prepared from an in vitro model of chlamydial persistence. Preparation of RNA/cDNA and details of the analyses are given in “Patients and methods”. Bars represent standard error.
Expression of selected C trachomatis orthologues during persistent infection in vivo
With one interesting exception, the human monocyte model of C trachomatis persistence has proved to be extremely accurate in reflecting the pattern of chlamydial gene expression in vivo in synovial tissues.8,9 That exception was the identification of a different transcript pattern from the chlamydial groEL gene (Ct110, encodes the authentic heat shock protein (hsp)60) between the monocyte model of persistence and synovial tissue samples from patients with Chlamydia associated arthritis10; we understood this observation to indicate that the cellular context in the synovium differs in important ways from that of pure monocytic cells in culture.
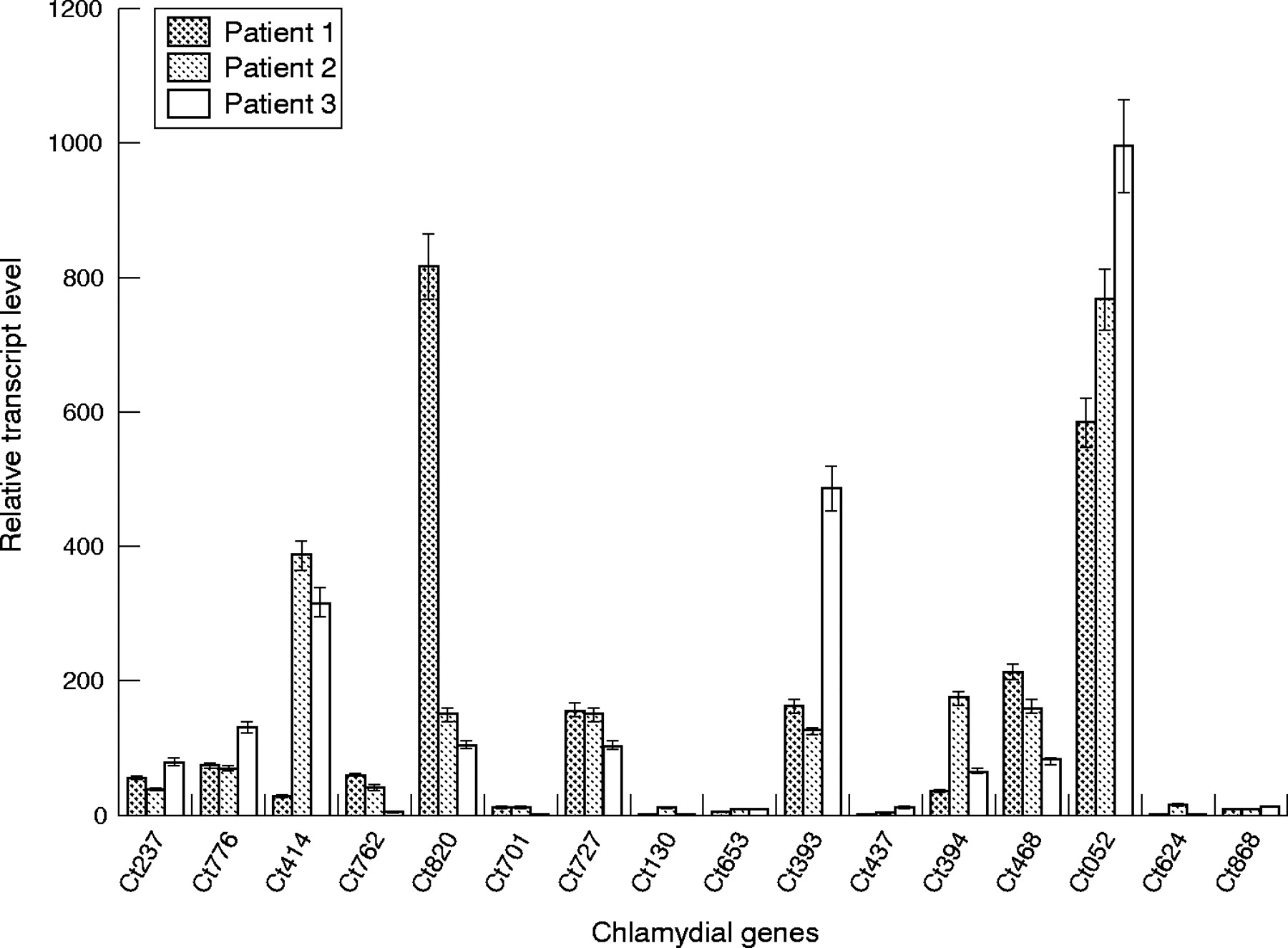
To determine whether in vivo and in vitro expression differed for any of the 16 tested chlamydial persistence orthologue genes, we repeated the real time RT-PCR assays using RNA/cDNA prepared from synovial biopsy specimens of three patients with arthritis known to be PCR positive for C trachomatis in that tissue. Figure 2 presents the results of those assays. The Ct820, Ct727, Ct393, and Ct052 genes all showed expression in these patient samples, as they did in the monocyte model of chlamydial persistence. However, the genes that displayed essentially no up regulation of expression in the monocyte system (Ct762, Ct701, Ct624, Ct868) all showed some level of increased expression in the patient samples, consistent with data from the mycobacterial study.

{kind=link}
{kind=link}
Real time RT-PCR analysis of transcript levels from 16 C trachomatis genes identified as orthologous to M tuberculosis genes required for in vivo growth and persistence, using cDNA prepared from Chlamydia infected synovial tissue samples. Preparation of RNA/cDNA and details of the analyses are given in “Patients and methods”. Bars represent standard error.
Ct414 (pmpC, polymorphic membrane protein), Ct394 (hrcA, transcriptional repressor), and Ct468 (pilR/atoC, sensor subunit of a two-component system) all showed similar levels of expression in one or more of the patient samples relative to that found in the monocyte model of persistence. Ct776 (aas, 2-acylglycerophosphoethanolamine acyltransferase) and Ct437 (fusA, elongation factor G protein) also were expressed at generally the same levels in patient samples as in the in vitro monocyte system. We assume that differences in transcript levels among the three patients for the genes assessed (for example, Ct414, Ct820) were a function of disease duration and genetic and other characteristics specific to each individual; these characteristics determine the details of host-pathogen interaction for each patient, in turn influencing the course of pathogenesis for each individual patient.
DISCUSSION
Molecular genetic and other observations have demonstrated that C trachomatis infecting synovial tissues in patients with Chlamydia associated arthritis are in the persistent, rather than the actively growing, state.2,3 Although we have some knowledge of the transcriptional and morphological characteristics of persistent Chlamydia, we have essentially no understanding of which specific aspects of host-pathogen interaction are responsible for eliciting chlamydial persistence in either the monocyte model system or in synovial materials. This lack of understanding results primarily from the absence of a system for genetic manipulation of any chlamydial species, and from the large number of genes specifying proteins of unknown function encoded by all chlamydial genomes so far sequenced. In the present report, we extrapolate to C trachomatis molecular genetic data from a persistence related study of M tuberculosis, an organism amenable to genetic manipulation and which undergoes persistent infection.17,22 We show that 35% of the 194 mycobacterial genes identified in the parent study as showing significant transcriptional up regulation in support of persistent infection have orthologous coding sequences on the C trachomatis genome. We further show that most of a selected set of those chlamydial orthologues do indeed show increased expression in an in vitro model of chlamydial persistence and in relevant patient samples, compared with their normal expression levels during active infection. Thus, the results presented here indicate that some molecular genetic details underlying chlamydial persistence can be gleaned or extrapolated from experimental work in other organisms. Most importantly, however, the data given add new details to the emerging picture of the genetic basis underlying persistent infection by C trachomatis, in both a relevant in vitro system and in vivo in samples from patients with arthritis.
The genome of M tuberculosis is about 4.4 mbp in length and encodes about 4000 genes.26 Most of the 194 genes identified in the persistence study of this organism are expressed at only low level or not at all during axenic growth in culture,22 suggesting that persistence for M tuberculosis may depend on expression of a particular set of genes devoted largely or exclusively to establishment of that state. In contrast, the C trachomatis genome specifies just over 900 coding sequences, and all of them are expressed at one time or another during active growth of the organism.15,16,24,25 This suggests that, unlike M tuberculosis, C trachomatis does not possess a gene or gene set whose sole function is the genesis and/or maintenance of the persistent infection state. Thus, while M tuberculosis persistence may derive largely from expression of a specific set of genes, in C trachomatis development of that state must be a function of readjustment of transcript levels from genes already being expressed. The challenge will be to understand whether, and if so by what means, that readjustment results from input from the host monocytic cell in the joint, and how that readjustment of gene expression alters chlamydial biochemistry and physiology.
It is difficult to deduce from the results presented here, and from those in the mycobacterial study, whether and if so how, metabolic processes are similarly modified overall between the actively growing and persistent infection states of these two bacterial pathogens. Although many of the M tuberculosis genes up regulated during in vivo growth and persistence have known functions, the majority of genes (107/194 (55%)) so identified encode proteins of unknown function.22 Nine of the C trachomatis orthologues of mycobacterial genes found in our BLAST search also were coding sequences specifying proteins of unknown function, but importantly, most of the unknown M tuberculosis genes in the persistence study were shown to be unique to that organism. As pointed out in the Sassetti and Rubin study, these observations indicate that mycobacteriae appear to have evolved mechanisms for in vivo survival and persistence that are unique—that is, not shared by other obligate or facultative intracellular pathogens. Similarly, most coding sequences specifying products of unknown function in the C trachomatis genome are either Chlamydia- (genus-) specific or specific to that organism, probably indicating that the genetic underpinnings of chlamydial persistence are essentially unique to this group as well. Indeed, we suspect that many critical molecular genetic and other details differ between these two organisms in their metabolic and other characteristics of persistence. None the less, while caution must be exercised in extrapolating the genetic mechanisms underlying persistence in one organism to those performing a similar function in another, circumspect exercise of such extrapolations can be useful.
The panel of M tuberculosis genes underlying in vivo growth and persistence, and the group of C trachomatis genes identified as orthologous to them, fall into the same general functional categories, with the exception that the former panel included more genes encoding lipid metabolism related products than did the latter. As mentioned, given the limited panel of chlamydial genes identified as being orthologous to mycobacterial persistence related genes, it is difficult to form a detailed picture of the overall transcript pattern underlying chlamydial persistence in monocytic cells, and the overall metabolic characteristics of persistence for the organisms that result from that gene expression pattern. One study provided a transcriptome analysis of chlamydial gene expression in an in vitro model of persistence which was different from the monocyte system used here.27 Collaborative studies between this laboratory and another have shown, however, that the profiles of gene expression differ importantly among the several currently employed in vitro model systems of persistence studied in various laboratories (Klos A, HCG, APH, unpublished observations). In the case of the monocyte model of chlamydial persistence, which we consider to be the most relevant for studies of pathogenesis in Chlamydia associated arthritis, understanding the transcriptional and metabolic modifications underlying persistence must derive from full transcriptome analyses of that system. We are now performing those analyses.
Acknowledgments
This work was supported by grants AR-42541 (APH), AR-47186 (HCG), and AI-44493 (JAW-H) from the US National Institutes of Health.
REFERENCES
Footnotes
-
Published Online First 28 September 2005
-
Competing interest: None.