Article Text

Abstract
Objective In anti-citrullinated protein antibody positive rheumatoid arthritis (ACPA-positive RA), a particular subset of HLA-DRB1 alleles, called shared epitope (SE) alleles, is a highly influential genetic risk factor. Here, we investigated whether non-HLA single nucleotide polymorphisms (SNP), conferring low disease risk on their own, interact with SE alleles more frequently than expected by chance and if such genetic interactions influence the HLA-DRB1 SE effect concerning risk to ACPA-positive RA.
Methods We computed the attributable proportion (AP) due to additive interaction at genome-wide level for two independent ACPA-positive RA cohorts: the Swedish epidemiological investigation of rheumatoid arthritis (EIRA) and the North American rheumatoid arthritis consortium (NARAC). Then, we tested for differences in the AP p value distributions observed for two groups of SNPs, non-associated and associated with disease. We also evaluated whether the SNPs in interaction with HLA-DRB1 were cis-eQTLs in the SE alleles context in peripheral blood mononuclear cells from patients with ACPA-positive RA (SE-eQTLs).
Results We found a strong enrichment of significant interactions (AP p<0.05) between the HLA-DRB1 SE alleles and the group of SNPs associated with ACPA-positive RA in both cohorts (Kolmogorov-Smirnov test D=0.35 for EIRA and D=0.25 for NARAC, p<2.2e-16 for both). Interestingly, 564 out of 1492 SNPs in consistent interaction for both cohorts were significant SE-eQTLs. Finally, we observed that the effect size of HLA-DRB1 SE alleles for disease decreases from 5.2 to 2.5 after removal of the risk alleles of the two top interacting SNPs (rs2476601 and rs10739581).
Conclusion Our data demonstrate that there are massive genetic interactions between the HLA-DRB1 SE alleles and non-HLA genetic variants in ACPA-positive RA.
- rheumatoid arthritis
- ANT-CCP
- gene polymorphism
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
In rheumatoid arthritis (RA (OMIM: 180300)), a particular subset of HLA-DRB1 gene variants (major alleles at *01, *04 and *10 groups), commonly called shared epitope (SE) alleles, is the most important genetic contributor for the risk of developing anti-citrullinated protein antibody (ACPA)-positive RA.1–3 It is noteworthy that the strength of the association between non-HLA genetic variants and ACPA-positive RA risk is, in general, very moderate in comparison to that of the HLA-DRB1 SE alleles4–7 (figure 1A). This prompted us to investigate whether the HLA-DRB1 SE alleles could be a genetic hub8 that captures multiple interactions. Indeed, previous studies have demonstrated interactions between the HLA-DRB1 SE alleles and several single nucleotide polymorphisms (SNP), including variations in PTPN22, HTR2A and MAP2K4 with regard to the risk of developing ACPA-positive RA,9–12 where the combination of both risk factors shows significantly higher risk (measured as OR) than the sum of their separate effects. Departure from additivity is a way to demonstrate interaction between risk factors regarding the risk of disease. The additive scale, defined by attributable proportion (AP) due to interaction, has the advantage of a straightforward interpretation in the sufficient-component cause model framework.9 13–16
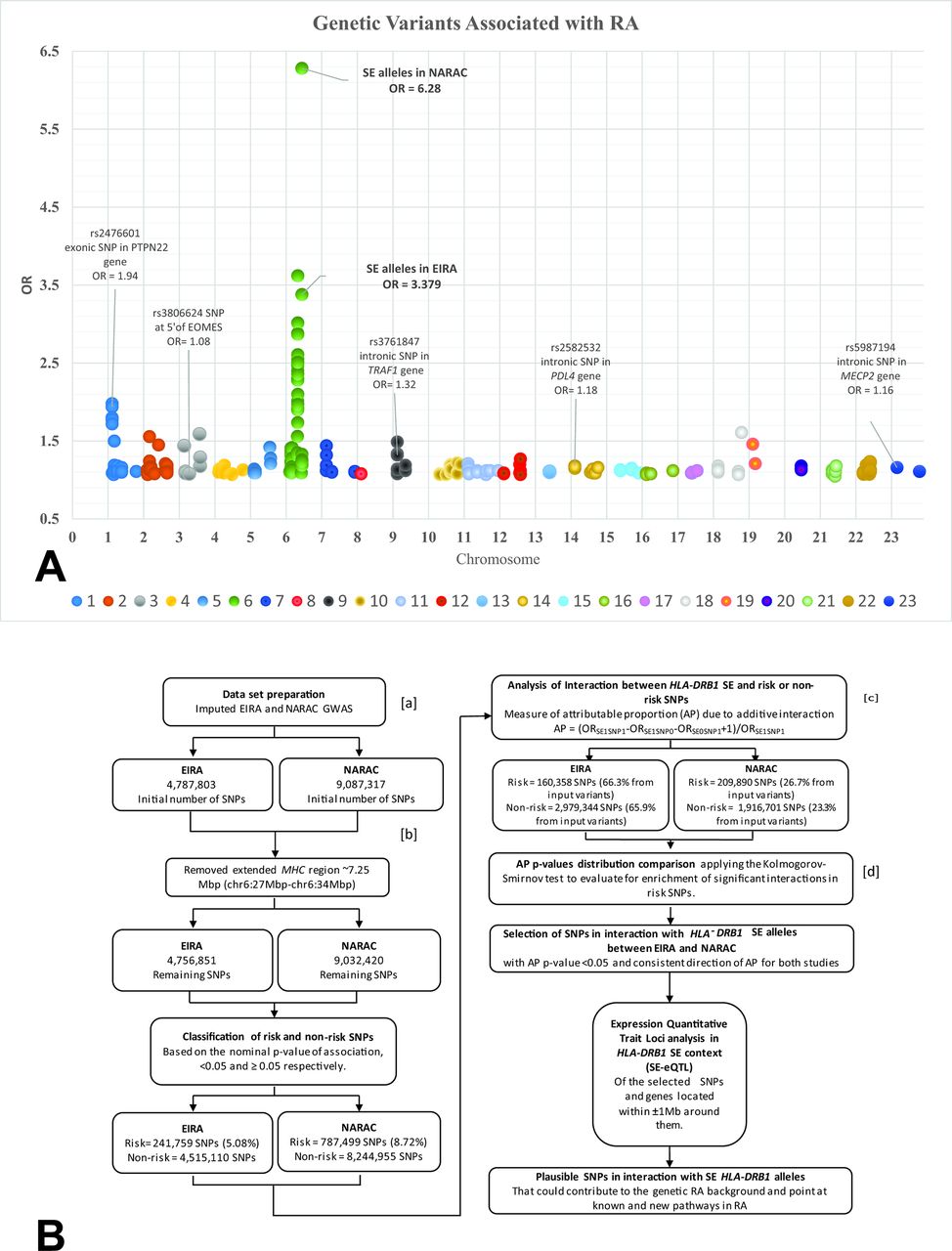
(A) Genetic variants associated with ACPA-positive RA. This plot represents the association signals (p<1.0e-05) from different GWAS in ACPA-positive RA, taken from the NHGRI-EBI GWAS catalogue (https://www.ebi.ac.uk/gwas/home).46–48 X-axis: genomic positions, including chromosome X (marked as 23). Y-axis: the OR value observed for each SNP in different studies. Some examples are pointed. (B) Methodology workflow. (a) The workflow was also applied with non-imputed genotyping data (online supplementary table S2). (b) An alternative step excluding the PTPN22 locus was included at this point. (c) The AP value, its respective p value and CI (95% CI) were assessed using logistic regression implemented in GEISA (https://github.com/menzzana/geisa).13 27 28 (d) The classification of risk and non-risk SNPs was permuted 10 000 times and each time the KS test was applied. The workflow was implemented until this step for each of the 1000 SE permuted variables, a lower number of permutations due to computational constrains. Both types of permutations showed that less than 5% of the KS test will exhibit a p value less than 2.2e-16, strongly indicating that differences in the AP p value distribution detected by the KS test from the original data are unlikely to be by chance. ACPA-positive RA, anti-citrullinated protein antibody positive rheumatoid arthritis; EBI, European Bioinformatics Institute; EIRA, epidemiological investigation of rheumatoid arthritis; GWAS, genome-wide association study; KS, Kolmogorov-Smirnov test; LD, linkage disequilibrium; MAF, minor allele frequency; MHC, major histocompatibility locus; NARAC, North American rheumatoid arthritis consortium; NHGRI, National Human Genome Research Institute; PCA, principal component analysis; SE, shared epitope; SE0SNP1, absence of the HLA-DRB1 SE alleles and presence of the risk allele from the SNP; SE1SNP0: presence of the HLA-DRB1 SE alleles and absence of the risk allele from the SNP; SE1SNP1, presence of the HLA-DRB1 SE alleles and the risk allele from the SNP; SNP, single nucleotide polymorphism. PTPN22 is abbreviation for the gene.
Supplemental material
In our current study, we aimed to investigate whether there is an enrichment of genetic interactions between non-HLA SNPs, conferring low disease risk on their own, and the major HLA-DRB1 related disease risk to develop ACPA-positive RA. We also explored to what extent the top interactions influence the association between HLA-DRB1 and risk to ACPA-positive RA. First, we assessed departure from additivity regarding the interaction between the HLA-DRB1 SE alleles and SNPs at the genome-wide level. The outcome of this analysis was used to investigate the potential enrichment of significant interactions among certain variants by comparing the distribution of the p value of interaction between two defined groups of SNPs: the pool of variants which exhibited a significant nominal association with ACPA-positive RA in comparison to SNPs that are not associated with disease risk. We thereafter performed a replication study with the same approach in an independent ACPA-positive RA cohort. Second, we performed an expression quantitative trait loci (eQTL) analysis for non-HLA SNPs in interaction with HLA-DRB1, stratified by the HLA-DRB1 SE alleles in order to identify genes influenced by the interactions. Finally, we analysed the effect size from the HLA-DRB1 SE alleles concerning the risk of developing ACPA-positive RA before and after step-by-step removal of the effect of the risk alleles of the strongest SNPs in interacting with HLA-DRB1 SE. Our observations indicated that the HLA-DRB1 SE alleles act as a hub of cumulative additive interactions with multiple genetic variants in the development of ACPA-positive RA. We proposed that the analytic approach used here provides a novel way to study the impact of gene–gene interactions and the role of HLA in other autoimmune diseases.
Materials and methods
The methodology workflow is represented in figure 1B.
Studied populations
This project was based on genome-wide association study (GWAS) data from two independent case–control studies of RA, epidemiological investigation of rheumatoid arthritis (EIRA)6 14 17–20 and North American rheumatoid arthritis consortium (NARAC).6 18 21 22 A total of 4291 individuals were included in this study, where 2018 are ACPA-positive RA cases and 2273 are healthy controls (table 1). A detailed description is found in the online supplementary information.
Supplemental material
Description of studied populations
Genotyping and data filtering
HLA typing was performed by sequence-specific primer PCR assay as described elsewhere.23 24 A standard quality control for GWAS was performed before and after imputation for both cohorts (see the description in the online supplementary information). The extended MHC region (chr6:27339429-34586722, GRCh37/hg19) was removed from the analyses, to exclude influence of the high linkage disequilibrium (LD) and independent signals of association with ACPA-positive RA in the locus. Association tests for EIRA and NARAC were applied using logistic regression model in plink (V.1.07).25 Based on the nominal p values of association, the SNPs were grouped into risk (p<0.05) or non-risk SNPs (p≥0.05) (figure 1B and table 2).
The Kolmogorov-Smirnov (KS) test for AP p value distributions of the interaction analysis with the HLA-DRB1 SE alleles and risk or non-risk SNPs in EIRA and NARAC imputed data
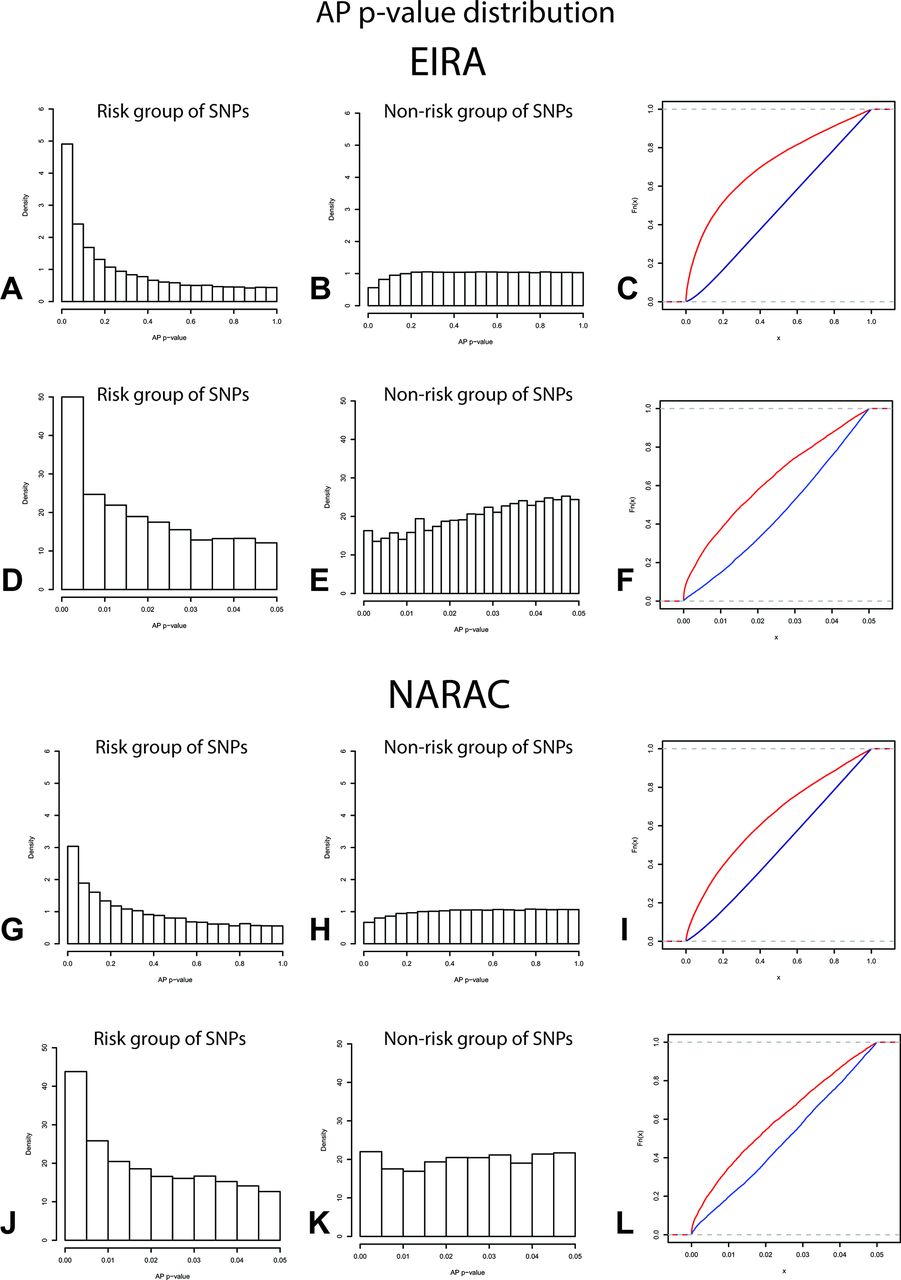
AP p value distributions for interaction between the HLA-DRB1 shared epitope (SE) alleles and two groups of genetic variants. (A, G) Density plots of AP p values for the interaction between the HLA-DRB1 SE alleles and the risk group of SNPs (nominal p value of association <0.05) or (B, H) non-risk group of SNPs (nominal p value of association ≥0.05) in the EIRA or NARAC studies, respectively. (C, I) The respective ECDF plots of the AP p values from risk (red line) or non-risk (blue line) SNPs in interaction with the HLA-DRB1 SE alleles in the respective studies (table 2). The AP p value distribution and the KS tests on the fraction of AP p values less than 0.05 are represented in panels (D)–(F) for EIRA study and (J)–(L) for NARAC study. AP, attributable proportion due to interaction; ECDF, empirical cumulative distribution function; EIRA, epidemiological investigation of rheumatoid arthritis; KS, Kolmogorov-Smirnov test; NARAC, North American rheumatoid arthritis consortium; SNP, single nucleotide polymorphism.
Interaction analysis
The additive model was used to test for interactions between the HLA-DRB1 SE alleles and each SNP from the EIRA and NARAC GWAS. The null hypothesis of the additive model assumes that there is additivity between the different sufficient causes for a phenotype, while the alternative hypothesis is assumed when departure from additivity is observed. The departure from additivity is estimated by the AP due to interaction using OR as measure of association,26 with the following equation:

where 1 and 0 refer to presence or absence of the risk factor/allele, respectively, the SE0SNP0 was used as a reference group. A cut-off of five observations for each of the cell frequencies was applied. The gender and the first 10 principal components (online supplementary information) were included as covariates in the model. The AP, its respective p value and CI (95% CI) were assessed using logistic regression implemented in GEISA (V.0.1.12)13 27 28 (table 2).
Comparison of the distribution of AP p values between the risk and non-risk groups of SNPs and quality controls
The distribution of AP p values observed in the interaction analysis from the ACPA-positive RA risk SNPs was compared with the distribution of AP p values observed from the non-risk SNPs using the Kolmogorov-Smirnov (KS) test, implemented in the stats package of R software (V.3.3.2).29 The KS test statistic quantifies the maximum distance (D) between the two empirical cumulative distribution functions of the AP p values from the risk and non-risk SNP groups.
The SNP category of risk and non-risk was permuted 10 000 times to evaluate whether the observed AP p value distribution with the original SNP classification remains with random SNP classification. Similarly, the HLA-DRB1 SE allele variable was iterated 1000 times with the same purpose. Both types of permutations showed that less than 5% of the KS tests will exhibit a p value under 2.2e-16 (online supplementary information). The rs4507692 SNP (chromosome 7) was used as a negative control instead of HLA-DRB1 SE alleles, since the rs4507692 SNP is not associated with RA but has the same minor allele frequency as the HLA-DRB1 SE alleles (table 1).
The SNPs from the PTPN22 locus (chr1:113679091-114679090, GRCh37/hg19) were removed to apply the same workflow, from step 1 to 9 in figure 1B, to determine the influence of this locus in the enrichment analysis, due to the known gene–gene interaction between the rs2476601variant (or SNPs in LD with this variant) and the HLA-DRB1 SE alleles.9 Additionally, to address possible inflation in the results due to imputation and LD, the same steps as described in figure 1B were implemented with (1) raw (non-imputed) GWAS data, (2) with the exclusion of all variants from chromosome 6, and (3) LD pruned GWAS data.
SNPs in interaction with SE between EIRA and NARAC
We selected those variants with AP p<0.05 and same AP direction from both the EIRA and NARAC studies to evaluate their distribution across the genome. False discovery rate (FDR) correction was applied to these SNPs with a 5% threshold (online supplementary table S3)
Conditional eQTL analysis in the context of the HLA-DRB1 SE alleles (SE-eQTL)
The selected SNPs were tested for eQTLs in the carrier and non-carriers of HLA-DRB1 SE alleles (SE-eQTLs) for genes±1 Mbp around the SNPs. Data from peripheral blood mononuclear cells (PBMC) of 97 patients with ACPA-positive RA (69% female) from the COMBINE study30 were included. The mixed-linear model function in the nlme 3.1 package from R/Bioconductor (V.3.3.2)31 was used for the analysis (details in the online supplementary information).
Results
Interaction between the ACPA-positive RA-associated SNPs and HLA-DRB1 SE alleles is more common than with non-associated SNPs
EIRA study was the discovery cohort to test for enrichment of significant interactions between the HLA-DRB1 SE alleles and the predefined risk SNPs from this study. The risk SNPs represent 5% of the variants analysed for interaction in EIRA. Out of these risk SNPs, 24.5% exhibited an AP p value less than 0.05 (table 2, figure 2A). On the other hand, among the non-risk variants (nominal p values of association ≥0.05) representing the remaining 95% of analysed SNPs, only 2.8% displayed a significant interaction (AP p<0.05) with the HLA-DRB1 SE alleles (table 2, figure 2B). This striking difference in the frequency of significant interactions is reflected in the KS test (D=0.35, p<2.2e-16) (table 2, figure 2C). The enrichment was also observed when only the segments of the AP p values below 0.05 for risk and non-risk SNPs were compared (KS test D=0.25, p<2.2e-16, figure 2D–F), suggesting that the enrichment of significant interactions corresponds mainly to the low AP p values of the risk group of SNPs.
Importantly, this enrichment of low AP p values in the risk variants is totally absent when the rs4507692 SNP was tested instead of the HLA-DRB1 SE variable as negative control. The proportion of interacting risk SNPs with the rs4507692 variant dropped to 2.8% (table 1 and online supplementary table S1 and figure S1a–f). Since the same group of risk variants was tested for interaction with the HLA-DRB1 SE alleles and rs4507692 SNP, both AP p value distributions were comparable (KS test D value=0.35, p<2.2e-16) (online supplementary figure S2a). This comparison confirmed that the enrichment of significant interactions is only present when the risk SNPs and the HLA-DRB1 SE alleles are tested.
Supplemental material
The enrichment of significant interactions was not remarkably affected after removing the PTPN22 locus (KS test D=0.353, p<2.2e-16), highlighting that the strong increment of significant interactions is due to multiple ACPA-positive RA risk variants.
Consistent enrichment of significant interactions was observed when the workflow was applied to non-imputed genotyping data for EIRA, before (KS test D=0.33, p<2.2e-16) and after (KS test D=0.33, p<2.2e-16) removing all the variants from chromosome 6 and LD pruning (online supplementary table S2). The enrichment of significant interactions in the risk group of SNPs was detected also when more stringent thresholds of nominal p value for association (0.005 and 0.0005) were used to classify associated SNPs (online supplementary figure S3).
Supplemental material
Altogether, the results from EIRA study indicate that there is a strong enrichment of significant interactions between non-HLA risk variants and HLA-DRB1 SE alleles in ACPA-positive RA.
An independent replication supports the observed enrichment of significant interactions between the ACPA-positive RA-associated SNPs and the HLA-DRB1 SE alleles
To confirm the results, the independent NARAC study was used. Consistently, a significant proportion of interactions with AP p values below 0.05 was detected between the HLA-DRB1 SE alleles and the risk SNPs (15.2%), compared with the ones found with the non-risk SNPs (3.3%) (KS test D=0.25, p<2.2e-16, table 2, figure 2G–I). This observation was corroborated when only the fraction of AP p values below 0.05 was compared between the risk and non-risk groups of variants in the NARAC study (KS test D=0.17, p<2.2e-16, figure 2J–L). As in EIRA, there was no enrichment of significant interactions in the risk group (2.6%) compared with the non-risk group (3%) of SNPs when the negative control (rs4507694) was implemented (online supplementary table S1 and figure S1g–l). Consequently, the proportion of relevant interactions remained higher between the risk SNPs and HLA-DRB1 SE alleles, when compared with the proportion of such interactions with the risk SNPs and the rs4507692 variant (KS test D=0.26, p<2.2e-16; online supplementary figure S2b). Analogous results to EIRA were observed when the workflow was applied to non-imputed GWAS in the NARAC study (online supplementary table S2).
Chromosomes 1 and 9 were highlighted by the top interactions with the HLA-DRB1 SE alleles
The comparison of results from EIRA and NARAC studies identified 1492 SNPs in interaction with the HLA-DRB1 SE alleles, with AP p<0.05 and the same direction of AP for both studies (figure 3A,B, online supplementary table S3). The signals within chromosomes were ranked based on the minimum AP p value, the maximum AP value and the percentage of these 1492 SNPs in interaction with the HLA-DRB1 SE alleles (online supplementary table S4). Chromosomes 1 and 9 reach the highest position for both studied cohorts (minimum AP p=4.3e-10 in EIRA and p=1.6e-08 in NARAC; online supplementary table S4). Specifically, the loci 9q33 and 1p13 contain the top SNPs in interaction with the HLA-DRB1 SE alleles (figure 3C,D,H,I and online supplementary table S3). Chromosomes 2, 7, 8 and 13 followed in the ranking when the results from both EIRA and NARAC were considered. The majority (84.6%) of these SNPs in interaction with the HLA-DRB1 SE alleles exhibited positive AP values, and most of them were under 0.5 (figure 3A,B).

Selected SNPs from both studied cohorts with AP p<0.05 and the same direction of AP. The circos plots for (A) the EIRA study and (B) the NARAC study represent the 1492 selected SNPs in additive interaction with the HLA-DRB1 SE alleles. The outermost track of the circos plots is the cytoband for 22 human chromosomes. The y-axis of the second track is the negative logarithm of the AP p values (additive interaction with the HLA-DRB1 SE alleles). In the third track, the y-axis corresponds to the AP value. The internal connector lines highlight the interactions that exhibited an AP p<1e-03. (C) and (D) panels represent the 1p13 locus centred on the rs2476601 SNP. This variant is a conditional eQTL in the HLA-DRB1 SE allele context for (E) PTPN22, (F) HIPK1 and (G) CSDE1 genes in PBMCs from the patients with ACPA-positive RA. (H) and (I) panels represent the 9q33 locus centred on the rs10739581 SNP. This variant is a conditional eQTL in the HLA-DRB1 SE allele context for (J) PSMD5 and (I) CDK5RAP2 genes. Panels (C), (D), (H) and (I) were done using LocusZoom (V.0.4.8) (http://locuszoom.org/genform.php?type=yourdata).49 AP, attributable proportion due to interaction; EIRA, epidemiological investigation of rheumatoid arthritis; FDR, false discovery rate; LD, linkage disequilibrium; NARAC, North American rheumatoid arthritis consortium; PBMC, peripheral blood mononuclear cell; SE, shared epitope; SE-eQTL, expression quantitative trait loci in shared epitope context; SNP, single nucleotide polymorphism. PTPN22, HIPK1, CSDE1, PSMD5 and CDK5RAP2 are abbreviations for the genes.
SNPs in interaction with the HLA-DRB1 SE alleles show functional features
The genotypes of 564 variants out of 1492 (37.7%) were considered to be SE-eQTLs in patients with ACPA-positive RA (online supplementary tables S5 and S6). The four top SE-eQTL pairs are graphically represented in the online supplementary figure S4.
Supplemental material
The non-synonymous variant rs2476601 in the PTPN22 gene (1p13) exhibited the most significant interaction with the HLA-DRB1 SE alleles (figure 3C,D). In turn, this polymorphism is an SE-eQTL for PTPN22 (protein tyrosine phosphatase, non-receptor type 22), HIPK1 (homeodomain interacting protein kinase 1) and CSDE1 (cold shock domain containing E1) genes (figure 3E–G, online supplementary table S5).
The rs10739581 SNP (9q33) is the second top replicated variant in interaction with HLA-DRB1 SE alleles (EIRA: AP=0.4, 95% CI 0.24 to 0.57, AP p=1.4e-6, FDR q=0.04; NARAC: AP=0.43, 95% CI 0.28 to 0.59, AP p=2.1e-8, FDR q=2.5e-4, online supplementary table S3). This variant is in high LD (r2>0.9) with the rs3761847 SNP that has previously been associated with RA.4 6 19 23 24 The rs10739581 SNP is an SE-eQTL for the CDK5RAP2 (CDK5 regulatory subunit associated protein 2) and PSMD5 (proteasome 26S subunit, non-ATPase 5) genes (figure 3J,K, online supplementary table S5).
These results suggest a plausible involvement of the 1492 selected SNPs in the pathogenesis of ACPA-positive RA through interaction with the HLA-DRB1 SE alleles.
The top interacting SNPs explain a considerable part of the influence from HLA-DRB1 SE on ACPA-positive RA
Finally, we observed that the step-by-step removal of the effect from the risk alleles of the rs2476601 and rs10739581, the two top replicated SNPs in interaction with the HLA-DRB1 SE alleles, decreases the effect size of SE alleles for ACPA-positive RA in the studied cohorts (figure 4). This observation clearly suggests that there is a strong mutual influence, reflected in the additive interactions detected, between non-HLA genetic variants and the HLA-DRB1 SE alleles in the development of ACPA-positive RA.

{kind=link}
{kind=link}
![[SP4.jpg]](https://ard.bmj.com/content/annrheumdis/77/10/1454/DC3/embed/inline-supplementary-material-3.jpg?download=true){kind=link}
![[SP5.jpg]](https://ard.bmj.com/content/annrheumdis/77/10/1454/DC4/embed/inline-supplementary-material-4.jpg?download=true){kind=link}
{kind=link}
![[SP6.jpg]](https://ard.bmj.com/content/annrheumdis/77/10/1454/DC5/embed/inline-supplementary-material-5.jpg?download=true){kind=link}
{kind=link}
Three-factor OR calculation, the HLA-DRB1 SE alleles and the top two SNPs in significant interaction. Panel (A) shows data from EIRA study while panel (B) shows data from NARAC study. The x-axis corresponds to the combinations of presence or absence of risk alleles from three factors in two models. The factors are the HLA-DRB1 SE alleles (presence—Y, or absence—N), the rs2476601 SNP (G>A, 1p13 locus) and the rs10739581 SNP (T>C, 9q33 locus). The rs2476601 and rs10739581 SNPs are in significant interaction with the HLA-DRB1 SE alleles after FDR correction in both EIRA (AP=0.45, 95% CI 0.31 to 0.60, p=4.3e-10, FDR q=5.2e-5 and AP=0.40, 95% CI 0.24 to 0.57, p=1.4e-6, FDR q=0.04, respectively) and NARAC (AP=0.41, 95% CI 0.23 to 0.6, p=1.1e-5, FDR q=0.04 and AP=0.43, 95% CI 0.28 to 0.6, p=2.1e-8, FDR q=2.5e-4, respectively). The y-axis represents the combined OR with 95% CI. AP, attributable proportion due to interaction; EIRA, epidemiological investigation of rheumatoid arthritis; FDR, false discovery rate; NARAC, North American rheumatoid arthritis consortium; SE, shared epitope; SNP, single nucleotide polymorphism.
Discussion
Our study demonstrates that there is an important enrichment of RA-associated SNPs interacting with HLA-DRB1 SE alleles concerning the risk to develop ACPA-positive RA. Importantly, there is a gradual decrease in the effect size of the HLA-DRB1 SE alleles in the risk of ACPA-positive RA after adjusting for top interacting SNPs. Based on these findings, we propose a concept called the dominion hypothesis, which suggests that the HLA-DRB1 SE alleles function as a genetic hub that is involved in multiple interactions with non-HLA genetic variants that by themselves have a modest effect size in RA. These interacting non-HLA variants do cumulatively contribute to the high effect size of the HLA-DRB1 SE alleles concerning risk to develop ACPA-positive RA. The dominion hypothesis has its foundations in the sufficient-component cause model,16 which suggests that several diverse components are part of a sufficient cause for a disease in a given affected individual, where each sufficient cause can include one or more component causes. Together, these component causes can form a minimal set of conditions that drive disease.32 Our hypothesis integrates the HLA alleles with other genetic variations across the human genome thereby potentially providing knowledge about the mechanisms behind this autoimmune disease.
The statistical approach in our study resulted in a list of 1492 SNPs (~270 independent loci, online supplementary table S7) as good candidates interacting with the HLA-DRB1 SE alleles in conferring risk for developing of ACPA-positive RA. Out of these SNPs 37.7% are SE-eQTLs that define genes that could be involved in the development of ACPA-positive RA. These findings suggest that the now identified additive interactions may reflect biological processes involved in the pathogenesis of ACPA-positive RA. For instance, the rs2476601 SNP appears to be an SE-eQTL for PTPN22, HIPK1 and CSDE1 genes in PBMCs of patients with ACPA-positive RA (figure 3 and online supplementary table S5). Furthermore, Capture Hi-C studies showed that the rs2476601 is in physical contact with the HIPK1 and CSDE1 genes in CD4+ and CD8+ T cells (https://www.chicp.org, data accessed: February 2018),33–35 which are relevant in RA.36 Another illustration is the rs1506691, rs6804917 and rs12630663 SNPs (online supplementary table S6) that are in physical contact in T cells with the EOMES gene (https://www.chicp.org),33 35 important in CD4+ T cell differentiation in the context of RA.37 Moreover, the HLA-DRB1 genotype appears to have a key role in shaping the T cell receptor repertoire.38 Our findings reinforce previous observations that have suggested that the HLA locus plays a special role in transgenomic regulatory mechanisms.39–41 The observed interactions might in some cases also reflect altered protein interactions in a given molecular pathway. For instance, it has been shown that the minor allele of rs2476601 SNP affects the LYP (protein encoded by PTPN22 gene)-CSK protein complex on the TCR signalling context,42 which in turn could have consequences for functions that involve TCR-peptide-MHC complexes. Additionally, one of the top interacting SNPs, the rs10739581, seems to be an SE-eQTL for the PSMD5 gene that is involved in antigen presentation and proteasome regulation.43 Interestingly, H3F3A and TNC genes that encode proteins that are known citrullinated autoantigens in RA44 45 are in the proximity of SNPs in interaction with HLA-DRB1 SE alleles (online supplementary table S6). Further interpretation and study of these interactions are required to understand their probable mechanisms in the RA pathogenesis.
When calculating the narrow heritability for ACPA-positive RA that could be explained by the current interaction analysis and the confirmed RA-associated loci,6 we obtained values of 54% for EIRA and 64% for NARAC (online supplementary information and supplementary table S8). However, these values should be considered cautiously since the calculation does not completely integrate interaction effects.
In conclusion, we used a systematic approach to investigate interactions at the genome-wide level between non-HLA and HLA-DRB1 alleles in ACPA-positive RA and we were able to identify a significant enrichment of interacting with HLA-DRB1 SE alleles among disease-associated non-HLA SNPs. We suggest that the dominion hypothesis might be used to explore the next level of complexity in this disease as well as in other multifactorial immune-mediated diseases that involve HLA.
Supplemental material
![[SP3.jpg]](https://ard.bmj.com/content/annrheumdis/77/10/1454/DC6/embed/inline-supplementary-material-6.jpg?download=true){kind=link}
Acknowledgments
We thank all the patients and control individuals involved in the EIRA, NARAC and COMBINE studies. Thanks to Magdalena Lindén, Tojo James and Ingrid Kockum who provided us an updated version of GEISA and good discussions about this tool. Thanks to Peter Gregersen and Soumya Raychouhundry who provided suggestions and data from NARAC. We thank Aaron Winkler for the scientific feedback and positive criticisms. We also thank the National Genomics Infrastructure (NGI) in Sweden for providing computational resource for our study and Meena Strömqvist for English language editing.
References
Footnotes
Handling editor Josef S Smolen
Twitter @LinMarDiaz
Contributors LMDG: conceptualisation, data curation, formal analysis, investigation, methodology, software, project administration, validation, visualisation, writing—original draft preparation and review and editing. DR: conceptualisation, formal analysis, methodology, resources, writing—review and editing. KS: conceptualisation, investigation, methodology, software, writing—review and editing. LF: conceptualisation, formal analysis, writing—review and editing. KC: conceptualisation, validation, writing—review and editing. BB: conceptualisation, writing—review and editing. SU: data curation. YO: data curation, resources, writing—review and editing. LA: conceptualisation, investigation, resources, software, writing—review and editing. LK: conceptualisation, funding acquisition, resources, writing—review and editing. LP: conceptualisation, funding acquisition, investigation, methodology, project administration, resources, supervision, validation, writing—original draft preparation and review and editing.
Funding This study was supported by the Swedish Council of Science (Vetenskapsrådet, https://www.vr.se/) (grant number 2015-03006); COMBINE project (Vinnova, https://www.vinnova.se/); BeTheCure EU IMI programme (http://cordis.europa.eu/project/rcn/203688_en.html); and Stiftelsen Konung Gustaf V:s 80-årsfond (KGV) Foundation (grant numbers FAI2014-0093, FAI2015-0207, FAI2016-0287, SGI2014-0022).
Competing interests None declared.
Patient consent Not required.
Ethics approval Karolinska Institutet Ethics Committee and the Regional Stockholm Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement The full output of the interaction analysis is available upon request. It is not included in the manuscript due to the size of the files.