Article Text

Abstract
Objectives The association between mutations in TNFAIP3, encoding the NF-kB regulatory protein A20, and a new autoinflammatory disease has recently been recognised. This study aims at describing the clinical phenotypes and disease course of patients with A20 haploinsufficiency (HA20).
Methods Data for all cases from the initial publication, and additional cases identified through collaborations since, were collected using standardised data collection forms.
Results A total of 16 patients (13 female) from seven families with a genetic diagnosis of HA20 were included. The disease commonly manifested in early childhood (range: first week of life to 29 years of age). The main clinical symptoms were recurrent oral, genital and/or gastrointestinal ulcers (16/16), musculoskeletal (9/16) and gastrointestinal complaints (9/16), cutaneous lesions (8/16), episodic fever (7/16), and recurrent infections (7/16). Clinical phenotypes varied considerably, even within families. Relapsing-remitting disease course was most common, and one patient died. Laboratory abnormalities included elevated acute-phase reactants and fluctuating presence of various autoantibodies such as antinuclear antibodies (4/10 patients tested) and anti-dsDNA (2/5). Tissue biopsy of different sites revealed non-specific chronic inflammation (6/12 patients tested), findings consistent with class V lupus nephritis in one patient, and pustules and normal results in two patients each. All patients were treated: 4/16 received colchicine and 12/16 various immunosuppressive agents. Cytokine inhibitors effectively suppressed systemic inflammation in 7/9 patients.
Conclusions Early-onset recurrent oral, genital and/or gastrointestinal ulcers are the hallmark feature of HA20. Frequency and intensity of other clinical manifestations varied highly. Treatment regimens should be based on disease severity, and cytokine inhibitors are often required to control relapses.
- autoinflammatory disease
- pediatric rheumatology
- ulcers
Statistics from Altmetric.com
Introduction
The protein A20, also known as TNAP3, encoded by TNFAIP3, plays a crucial role in the negative regulation of inflammation and immunity.1 TNFAIP3 is an ubiquitin-editing (deubiquitinase; DUB) enzyme with a critical function in the inhibition of key proinflammatory molecules, including inhibitor of nuclear factor kappa B kinase subunit gamma (IKKγ (NEMO)) and receptor-interacting protein kinase 1 (RIPK1), in the canonical NF-kB and other signalling pathways.2 Zhou and colleagues3 recently described a new autoinflammatory disease caused by heterozygous loss-of-function mutations in TNFAIP3, leading to haploinsufficiency of A20 (HA20). These mutations cause insufficient DUB activity of A20 and lead to increased NF-κB signalling and phosphorylation of c-Jun N-terminal kinase and p38 mitogen-activated protein kinases (MAPKs). HA20-associated mutations were found in six unrelated families who presented with mostly childhood-onset systemic inflammation and a ‘Behçet-like’ disorder that may lead to end-organ damage and death. Since the initial description, a few additional cases of HA20 have been reported in the literature.4–7 To date, the clinical manifestations, severity of symptoms, disease course and complications of this newly described disorder are not well described and appreciated. Early recognition and diagnosis are crucial, as targeted therapies may alter disease course and improve outcome.
Therefore, the aims of the study were (1) to describe the disease features and course, treatment regimens, complications and outcomes of patients; and (2) to improve clinical recognition of this poorly defined disorder.
Patients and methods
Sixteen patients from six unrelated families previously identified and diagnosed with HA20 by Zhou and colleagues3 at the National Institutes of Health in the USA were initially included in the study. The patients were followed at various centres worldwide and their charts were retrospectively reviewed by their primary care physicians. Because the spectrum of clinical phenotypes associated with HA20 is still widely unknown, these patients’ histories were used to prepare a comprehensive data collection form including all signs and symptoms thought to be related to HA20. In the second step, patient charts were again reviewed by the primary care physicians using the newly created data collection forms in order to collect detailed information. Demographic, clinical, laboratory, imaging and histological features were recorded. Disease course, treatment regimens, and disease- and treatment-related complications were captured. Additional cases were sought and one identified subsequently through collaborations.
All data were compiled and analysed using descriptive statistics. Patient consent, or consent from a parent in the case of children, was obtained by the responsible physician in the respective institutions.
Results
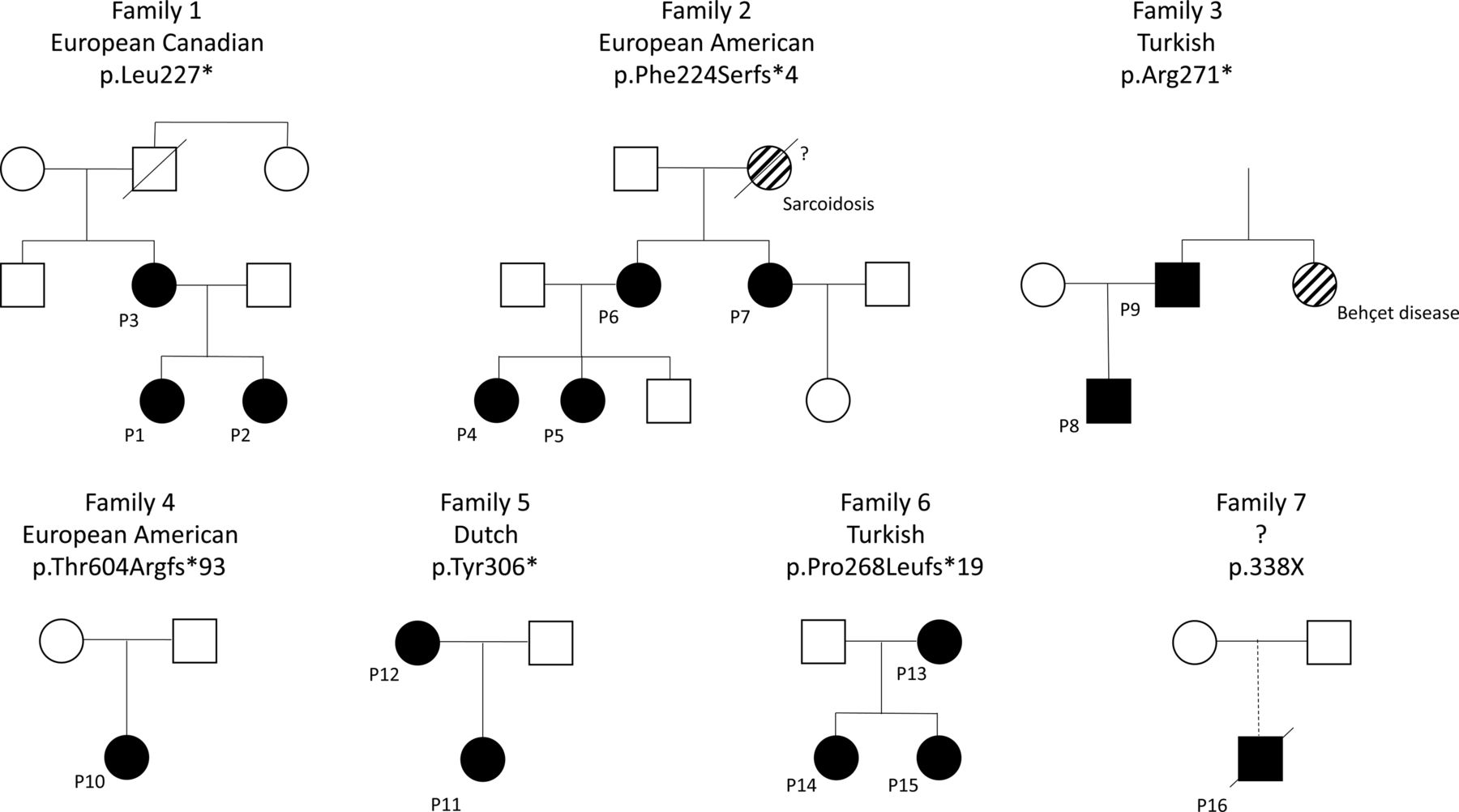
A total of 16 patients (81% female) from seven families were included (figure 1 and table 1). All patients were Caucasian, while the ethnic origin of patient 16 is unknown (figure 1). No consanguinity was reported within the families. Prior to the recognition of HA20, patients were diagnosed with various conditions including Behçet disease (BD), systemic lupus erythematosus (SLE), juvenile idiopathic arthritis (JIA), periodic fever, aphthous stomatitis, pharyngitis and adenitis (PFAPA), and inflammatory bowel disease (IBD). The grandmother of family 2 was diagnosed with sarcoidosis and succumbed to the complications of the disease. Given her medical history she is presumed to carry the F224Sfs*4 mutation present in four affected family members; however, her DNA sample was not available for genotyping. Thus, the molecular diagnosis of HA20 has not been confirmed and this patient was not included in this analysis.

Pedigrees of the seven families with mutations in TNFAIP3 leading to haploinsufficiency of A20 (HA20). The grandmother of family 2 was diagnosed with sarcoidosis and is thought to be affected by HA20, however died years prior to availability of genotyping. Patient 16 was adopted, and the ethnic origin is unknown.
Characteristics of patients with A20 haploinsufficiency
The demographic features of the patients are summarised in table 1.
Disease presentation
All patients were symptomatic and reduced penetrance was not reported in any family members. First symptoms occurred early, before 10 years of age in 11/16 (69%) patients, and disease onset ranged from the first week of life to 29 years of age. Clinical presentation was heterogeneous between families and between family members with the same mutation (figure 1 and table 2). In 10/16 (63%) patients, recurrent oral and/or genital ulcers were the symptoms leading to initial specialised medical consultation.
Clinical features of the patients with HA20 haploinsufficiency
Disease course
During disease course, symptoms and severity were highly variable. Clinical features emerged over a period of several years. All patients developed recurrent painful oral, genital and/or gastrointestinal ulcers. Other common symptoms that occurred at various time points during disease course included gastrointestinal complaints (9/16, 56%), polyarthritis and/or arthralgia (9/16, 56%), skin involvement (8/16, 50%) and recurrent fever (8/16, 50%). Less frequently, ocular (3/16, 19%) and cardiovascular involvement (3/16, 19%) was observed.
Seven patients (44%) suffered from recurrent, predominantly respiratory tract and otorhinolaryngological infections, especially during childhood. Infections were of viral and/or bacterial origin; only one of these patients was concomitantly treated with immunosuppressive agents. An overview of clinical symptoms occurring during the disease course is presented in tables 2 and 3.
Overview of clinical and laboratory characteristics observed in patients with A20 haploinsufficiency
Ulcers
Recurrent painful oral, genital and/or gastrointestinal ulcers were the hallmark feature of the disease. All 16 (100%) patients developed oral ulcers, whereas genital ulcers were observed in 15/16 (94%) and gastrointestinal ulcers in 6/9 patients with gastrointestinal complaints. Ulcerations recurred frequently (every month to every few months, at various intervals), isolated or in association with other symptoms such as fever, abdominal pain and arthralgia. Singular or multiple ulcers of various sizes (0.5–2 cm) lasted about 7–10 days and some healed with scarring (oral, genital or gastrointestinal location). Oral ulceration sites included the lips, tongue, cheeks, gums and hard palate, genital ulcers developed on vulva, vagina or the scrotum, and intestinal ulcerations were observed from the oesophagus to the rectum and perineum. Most patients could not identify an underlying trigger; in one patient, oral ulcers were exacerbated by acidic food.
Other clinical manifestations
Gastrointestinal symptoms were documented in 9/16 (56%) patients and ranged from isolated abdominal pain to severe intestinal inflammation with bowel perforation. Six (38%) patients suffered from recurrent, intermittently bloody diarrhoea. Musculoskeletal symptoms were frequent (9/16, 56%): polyarthritis was documented in seven patients, and in three of them arthritis was the initial disease manifestation. Cutaneous involvement (8/16, 50%) varied considerably and included pustular, folliculitis-like rashes, acne and dermal abscesses (figure 2). Ocular findings included severe, treatment-refractory anterior uveitis in two sisters (patients 1 and 2), as well as retinal vasculitis with chorioretinal scarring and macular fibrosis and anterior uveitis in another young girl (patient 4). Cardiovascular involvement was noted in three (19%) patients. Patient 8 was presented at age 9 months with fever and a pericardial effusion, which responded well to wide-spectrum antibiotic treatment. Another patient was diagnosed with pericarditis with a large effusion possibly related to Mycoplasma pneumoniae infection, which relapsed after discontinuation of corticosteroids but responded to colchicine (patient 10). In addition, patient 16, an 8-year-old boy with negative antiphospholipid antibodies and unremarkable hypercoagulability screen, presented with venous thrombi at sites of previous indwelling catheters. Two months later he was diagnosed with pulmonary arterial embolisms while he had no apparent venous thrombi. It is unclear whether these were true pulmonary embolisms or thromboses resulting from pulmonary artery vasculitis. Notably, none of the other patients developed thromboses or emboli. Neurological manifestations were reported in two (13%) patients, and both were diagnosed with central nervous system (CNS) vasculitis (patient 4 based on brain imaging and patient 16 based on a frontal lobe punctate). The clinical manifestations of the individual patients are shown in table 2.

{kind=link}
{kind=link}
Clinical and histological manifestations in patients with A20 haploinsufficiency (HA20). (A) Pustules and vesicles at various stages of development are seen. There is a mild desquamation and some hyperkeratosis. (B) Magnification image (20×) of the palmar pustule seen in (A), stained with H&E. An infiltrate containing lymphocytes and neutrophils is noted in the stratum corneum. (C) The colloidal iron staining for mucin (blue, magnification image 20×) shows extensive intradermal mucin accumulation. Mucin accumulation is a feature of connective tissue diseases, such as systemic lupus erythematosus, but is not characteristically seen in palmoplantar pustulosis or pustular psoriasis. This is an unusual finding and fits with the HA20 phenotype of both autoimmune and autoinflammatory manifestation. (D) Cutaneous ulcers on the buttock.
Laboratory and histology findings
Acute-phase reactants were elevated, especially during relapses. Most patients had normal acute-phase reactants in between flares; in three patients, C reactive protein and erythrocyte sedimentation rate were persistently elevated prior to treatment response. We observed fluctuating levels of various low-titre antibodies, including antinuclear antibodies (4/10 patients tested), anti-dsDNA (2/6) and anti-Sm/RNP (2/4) antibodies. Lupus anticoagulant was positive in 6/7 patients tested and anticardiolipin antibody in 2/5 patients. Patients 11 and 12 (family 5), both of whom suffered from recurrent viral and bacterial infections, were diagnosed with unclassified immunodeficiency with IgG subclass deficiency, absent polysaccharide vaccination response and lymphopaenia. None of the eight patients investigated for recurrent genital ulcers had evidence of herpes simplex infection.
Given clinical similarity with BD, we reviewed pathergy results and found they were variable among the 10 patients tested: positive in three patients, negative in six patients and inconclusive in one patient. HLA-B51 was positive in 2/5 patients tested. Tissue biopsy of at least one site was performed in 12/16 (75%) patients; pathological findings on histology included non-specific chronic inflammation in six, findings consistent with pustules and normal results in two patients each (bone marrow aspirate and intestinal mucosa in one, and lymph node in another) (figure 2). A kidney biopsy performed in patient 5 was consistent with class V lupus nephritis with glomerular basement membrane thickening, minimal inflammatory cell infiltrate, and extensive deposition of complement and immunoglobulins. Except for the frontal lobe punctate performed in patient 16, none of the tissue samples showed evidence of vasculitis. Online supplementary table 1 summarises the laboratory and histopathological features of patients with HA20.
Supplementary file 1
Treatment
All patients required treatment. Four patients responded well to monotherapy with colchicine. The other patients were treated with a monotherapy or a combination of immunosuppressive drugs, including systemic corticosteroids, disease-modifying drugs and cytokine inhibitors (anti-tumour necrosis factor (anti-TNF), anti-interleukin-1 (IL-1), anti-IL-6). After treatment-refractory disease courses, 12 patients eventually improved on either infliximab, anakinra, tofacitinib, colchicine or methotrexate in combination with thalidomide. More recently, therapeutic approaches were based on functional cytokine studies; cytokine inhibitors such as infliximab and anakinra proved effective in suppressing systemic inflammation in 7/9 patients. Most patients responded to high-dose corticosteroids but also suffered from major side effects. Patient 4 eventually underwent autologous haematopoietic stem cell transplantation for CNS vasculitis associated with a severe, SLE-resembling condition. She went into remission for 18 months, but subsequently developed a CNS vasculitis flare, anterior uveitis, idiopathic thrombocytopaenic purpura and recurrence of orogenital ulcers, for which various immunosuppressive agents were reinitiated. Previous and current treatment regimens of the individual patients are presented in table 1.
Three out of seven patients with recurrent respiratory tract and otorhinolaryngological infections underwent tonsillectomy; two patients (mother and daughter of family 5) received immunoglobulin replacement for low IgG subclass and the daughter also had repeat tympanostomy.
Outcome/complications
HA20 disease was characterised by unprovoked episodes of inflammatory symptoms or chronic inflammation. None of the patients developed lymphoma or malignancy. One patient died from HA20. Patient 16, who presented with severe intestinal involvement and presumed small vessel CNS vasculitis, died from uncontrollable disease and upper airway obstruction due to haemorrhage following erosion of the carotid artery from extension of bilateral tonsillar ulcerations. He was anticoagulated for pulmonary arterial embolisms.
Treatment-associated complications included corticosteroid-induced side effects such as Cushing syndrome, growth retardation, vertebral compression fractures, diabetes mellitus and arterial hypertension in five patients, and severe lymphopaenia and neutropaenia under azathioprine in another patient. Disease-associated and treatment-associated complications are shown in table 2.
Discussion
Herein, we describe the clinical manifestations and disease course of 16 patients with HA20, the largest cohort to date. The disease was characterised by early-onset systemic inflammation accompanied with recurrent oral, genital and/or gastrointestinal ulcers. Other clinical manifestations and disease course varied considerably even among patients with the same mutation, and ranged from severe or fatal multisystemic inflammation to mild disease with recurrent orogenital ulcers, arthralgia and cutaneous lesions. This suggests a role of modifying alleles in the disease progression and possible contribution of environmental factors such as diet.
Recurrent painful oral, genital and/or gastrointestinal ulcers were the hallmark feature in all subjects. Besides ulcers, various other, BD-like clinical manifestations such as articular, gastrointestinal, cutaneous and ocular symptoms were notable.8–13 As a consequence, the majority of patients (>70%) were initially diagnosed or suspected of having BD. HA20 is considered a monogenic type of BD due to highly penetrant novel germline mutations in TNFAIP3 and earlier onset symptoms. Polygenic and common BD, on the other hand, typically manifests in early adulthood and does not have clear dominant inheritance. Large case series of patients with paediatric BD reported a symptom onset in later childhood, between 6.9 and 12.3 years of age14–18; however, it is not clear how many of these patients carry mutations in TNFAIP3. Despite some similarities with BD, we recognised several important characteristics that help differentiate HA20 from polygenic BD, as shown in table 4. These other distinguishing observations in patients with HA20 included scarring oral ulcers, isolated anterior uveitis or retinal vasculitis with necrotising inflammation, recurrent fever, severe intestinal inflammation, elevated acute-phase reactants, the fluctuating presence of various autoantibodies, and a disease course refractory to standard treatment, all of which are unusual findings in typical BD.13 19
Clinical and laboratory features that are helpful to differentiate between A20 haploinsufficiency (HA20) and Behçet disease.
Although BD was the most common initial diagnosis, the heterogeneity of clinical phenotypes, variable temporal occurrence of symptoms, presence of various autoantibodies and histology resulted in the diagnosis of other inflammatory and autoimmune diseases. Family 1 presented with a clinical picture resembling JIA and/or rheumatoid arthritis (RA), patient 10 with symptoms compatible with PFAPA and patient 16 with features resembling Crohn’s disease. Half of the patients were found to have fluctuating autoantibodies and two sisters (patients 4 and 5) were initially diagnosed with SLE. Thus, it is likely that some patients with early-onset SLE might have mutations in TNFAIP3.
In addition to the 16 patients described in this study, four case reports including a total of 11 patients from four unrelated families were published in the literature since the initial publication by Zhou and colleagues.4–7 HA20 was recently reported in two Japanese families with an early-onset BD-like clinical picture.4 5 Similar to most patients in our cohort, these two families presented with recurrent orogenital ulcers and fevers; some family members also suffered from intestinal involvement, cutaneous lesions, nephrotic syndrome, polyarthritis or uveitis.4 5 Furthermore, HA20 was reported in a Japanese patient diagnosed with autoimmune lymphoproliferative syndrome (ALPS) and in a British boy with complex autoimmunity.6 7 The Japanese patient had presented with early onset, recurrent fever, bilateral cervical lymphadenopathy, hepatosplenomegaly and an extensive cutaneous rash suggestive of Kawasaki disease. Consistent with the diagnosis of ALPS, immunophenotyping revealed an increased percentage of double-negative T cells and a decrease in IgM memory B cells. However, unlike in patients with ALPS, the central memory, naïve, terminally differentiated effector memory T cells re-expressing CD45RA+, and effector memory subpopulations of CD3+ CD8+ T cells were normal in this patient.6 The boy of British ancestry was investigated for a complex, treatment-refractory autoimmune syndrome characterised by insulin-dependent diabetes, cytopaenias, hepatitis, enteropathy and interstitial lung disease.7 Prior to his molecular diagnosis of HA20, he underwent haematopoietic stem cell transplantation and is in complete remission (except for diabetes). He was diagnosed with a novel de novo heterozygous 2 bp deletion in TNFAIP3, p.V489Afs*7 in the second zinc finger domain (ZnF2). His disease-associated variant resides in a different domain of A20 from pathogenic mutations reported in all other patients and the ZnF4 domain may have other unappreciated functions.3 7 Most patients reported in our study are carriers for loss-of function protein truncating mutations in the ovarian tumour domain. This resulted in decreased deubiquitination of key signalling molecules, such as RIP1 and NEMO, increased activity of the NF-kB pathway, and high expression of proinflammatory cytokines.3 20 Although the molecular aetiologies of the British boy’s disease and our patients with HA20 are the same, his distinct clinical symptoms could be related to the presence of other unknown modifying gene alleles. Identification of additional HA20-associated mutations is necessary to better understand the full spectrum of phenotypes associated with the distribution of pathogenic mutations in A20.
Polymorphisms or mutations in TNFAIP3 have been associated with many autoimmune diseases, among them JIA, RA, IBD, SLE, type 1 diabetes, psoriasis, coeliac disease and coronary artery disease.20–27 In murine models, cell-specific ablation of A20 causes clinical features characteristic of these human diseases.1 20 Mice with A20 deficiency in myeloid cells develop polyarthritis mimicking human RA,28 while enterocyte-specific deficiency of A20 increases the susceptibility of mice to intestinal inflammation.29 Tissue-specific deletion of A20 in B cells or dendritic cells leads to the production of autoantibodies and an autoimmune syndrome resembling SLE.30 31 Mice deficient for A20 (A20−/−) develop severe multiorgan inflammation, cachexia and early death.32 Although A20 was initially described as required for termination of TNF-induced signals, the excessive inflammation observed in double-deficient mice, A20-TNF or A20-TNFR1, suggested that A20 might be critical for the regulation of TNF-independent signals. In addition, A20 has been shown to downregulate the activity of NLRP3 inflammasome and patients with HA20 had increased activity of NLRP3.3 A20 functions as a tumour suppressive gene and somatic mutations have been identified in B cell lymphomas.33 None of the patients in this cohort developed lymphoma or malignancy.
In clinical practice, HA20 may be considered in patients with an early-onset, dominantly inherited inflammatory disease who present with recurrent oral and genital ulcerations, fluctuating autoantibodies, and a treatment-refractory disease course.
There was no standardised treatment in this HA20 cohort. The patients received various immunosuppressive drugs prior to the diagnosis of HA20; more recently, therapeutic approaches were guided by functional cytokine studies. Elevated levels of many proinflammatory cytokines (IL-1, TNF, IL-6, IL-18, IFNγ, IP-10) have been documented in patients with HA20.3 20 Anticytokine agents such as anti-TNF or anti-IL-1 have been effective in suppressing the systemic inflammation in most of our patients. Haematopoietic stem cell transplant might be considered in patients with severe and treatment-refractory disease.
This study is limited by its retrospective design and other biases related to which patients received testing.3 Recurrent ulcers and a Behçet-like disease were characteristic features in this cohort, likely reflecting bias in which patients are screened for HA20. However, ulcers of variable extension and severity were documented in index cases and in all affected family members, suggesting that ulcers may be the hallmark feature of the disease. Given the retrospective study design and the disease pleiotropy, it is unlikely that HA20 is the unique underlying cause for all disease manifestations. The increased use of diagnostic whole exome sequencing will help identify other contributing disease modifying rare or common variants.34
In conclusion, HA20 disease in this cohort was characterised by early-onset inflammation and recurrent oral, genital and/or gastrointestinal ulcers and often positive family history. Other disease features and disease course varied considerably with an overall high morbidity and mortality. Treatment regimens should be based on severity of inflammatory manifestations, and often consist of targeted therapy with cytokine inhibitors to control the inflammation. In the future, identification of patients with monogenic inflammatory diseases will be important to understand the pathophysiology and guide treatments for common rheumatological diseases.
Acknowledgments
We thank the patients and their families for participating in this study. We thank Ingrid Goh for the technical support. The authors acknowledge Dres Chyi-Chia R Lee and Ed Cowen for their valuable assistance in the management of the cases.
References
Footnotes
Handling editor Josef S Smolen
Contributors FAA and RML: coordination of the study, data collection and analysis, manuscript preparation. EDB, SWC, EG, AG, PH, HLL, SO, DMS, DLS, AvR-K, IA and DLK: data collection and analysis, revision of the manuscript for important intellectual content. All authors approved the final version to be published.
Funding FAA was supported by grants from the Rhyner-Bangerter Foundation, Starr-Foundation, Swiss League against Rheumatism, Foundation W!, Alberta Children’s Hospital Research Institute Foundation and SickKids Foundation.
Competing interests None declared.
Ethics approval The study was approved by the Institutional Research Ethics Board (REB 1000053697).
Provenance and peer review Not commissioned; externally peer reviewed.