Article Text

Abstract
Objectives The genetic aetiology of osteoarthritis has not yet been elucidated. To enable a well-powered genome-wide association study (GWAS) for osteoarthritis, the authors have formed the arcOGEN Consortium, a UK-wide collaborative effort aiming to scan genome-wide over 7500 osteoarthritis cases in a two-stage genome-wide association scan. Here the authors report the findings of the stage 1 interim analysis.
Methods The authors have performed a genome-wide association scan for knee and hip osteoarthritis in 3177 cases and 4894 population-based controls from the UK. Replication of promising signals was carried out in silico in five further scans (44 449 individuals), and de novo in 14 534 independent samples, all of European descent.
Results None of the association signals the authors identified reach genome-wide levels of statistical significance, therefore stressing the need for corroboration in sample sets of a larger size. Application of analytical approaches to examine the allelic architecture of disease to the stage 1 genome-wide association scan data suggests that osteoarthritis is a highly polygenic disease with multiple risk variants conferring small effects.
Conclusions Identifying loci conferring susceptibility to osteoarthritis will require large-scale sample sizes and well-defined phenotypes to minimise heterogeneity.
This paper is freely available online under the BMJ Journals unlocked scheme, see http://ard.bmj.com/info/unlocked.dtl
Statistics from Altmetric.com
Osteoarthritis is the most common form of arthritis affecting 40% of people over the age of 70 years, and is associated with a substantial health economic burden. Osteoarthritis is thought to be caused by a complex interplay between environmental and genetic factors.1 As with many common complex disorders, the genetic architecture of osteoarthritis has not yet been characterised. Over the past decade, candidate gene association studies and genome-wide linkage scans have failed to identify robustly replicating osteoarthritis loci, with the notable exception of rs143383 in the GDF5 gene.2 A genome-wide association study (GWAS) recently reported a single novel locus associated with radiographically defined knee and/or hand osteoarthritis on chromosome 7q22 (rs3815148 in the COG5 gene),3 subsequently corroborated by a genome-wide association scan across four studies including the discovery set.4 Both established that osteoarthritis loci are represented by common variants (>0.2 minor-allele frequency), have small effect sizes (allelic OR ∼1.15), and reach genome-wide significance (p<5×10−8).4 5 These associations have characteristics typical of common complex traits and require large sample sizes for their detection. To enable a well-powered GWAS for osteoarthritis, we formed the arcOGEN Consortium, a UK-wide collaborative effort aiming to scan genome-wide over 7500 osteoarthritis cases in a two-stage genome-wide association scan. Here we report the findings of our GWAS stage 1 analysis and replication studies, and describe the outcomes of statistical analyses designed to model the genetic architecture of osteoarthritis.
Methods
An expanded description of the methods is provided in supplementary methods (available online only). The stage 1 genome-wide association scan included 3177 knee and/or hip osteoarthritis cases from the UK ascertained based on radiographic evidence of disease (Kellgren–Lawrence grade ≥2)6 or clinical evidence of disease to a level requiring joint replacement. Cases were genotyped using the Illumina Human610 platform (Illumina, San Diego, California, USA). We used 4894 early-access publicly available population-based UK controls from the Wellcome Trust Case Control Consortium 2 (WTCCC2) study, genotyped on the Illumina 1.2M Duo platform (Illumina). We compared allele frequencies across 514 898 autosomal single-nucleotide polymorphisms (SNP) passing quality control criteria. We also carried out joint-specific stratified analyses (for hip and knee osteoarthritis). We carried out sensitivity analyses by comparing genome-wide case genotypes against different control sets (one overlapping but typed on a different platform and one non-overlapping set of osteoarthritis-free ‘supercontrols’). We took forward 102 independent (r2<0.4) SNP with p<0.0001 to in-silico replication in three further osteoarthritis genome-wide association scan (from the deCODE, Framingham and Rotterdam studies) and a subset of 52 SNP in a UK-based genome-wide association scan (TwinsUK; supplementary table 1, available online only) (across 4124 cases and 37 581 controls in total). Based on meta-analysis results across arcOGEN and these in-silico replication datasets (supplementary results, available online only), we prioritised 36 SNP for de-novo replication in a further set of 6188 osteoarthritis cases and 8280 controls of European descent and for in-silico replication in 213 cases and 2531 controls from Estonia (supplementary table 1, available online only). To enhance our understanding of the genetic architecture of osteoarthritis, we applied analytical approaches7 to stage 1 arcOGEN data to test the theory of polygenic inheritance. We used the arcOGEN stage 1 GWAS to derive a set of independent (r2<0.05) associated SNP (∼62 000) from a subset of the data and then used this score allele set to evaluate the proportion of case–control status accounted for in the remaining samples.
Results
In our stage 1 genome-wide association scan analysis, we observed a slight excess of associations compared with the null distribution (supplementary figure 2, available online only) and similar patterns of association for the joint and gender-stratified analyses (supplementary results; supplementary figure 3, available online only). The genomic control inflation factor λ was 1.077, in keeping with other UK-based GWAS.8 Although there was no signal exceeding genome-wide significance (p<5×10−8), 89 SNP reached p values of less than 10−4 (as opposed to 51 expected under the null, binomial p=10−6).
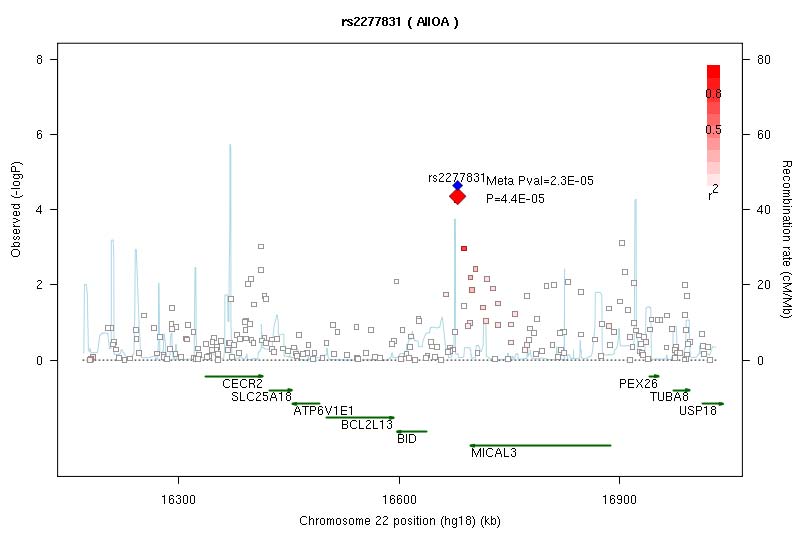
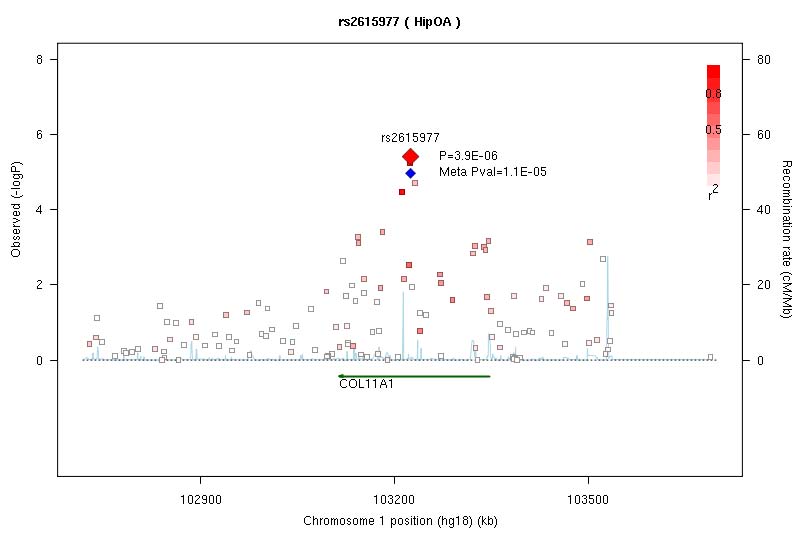
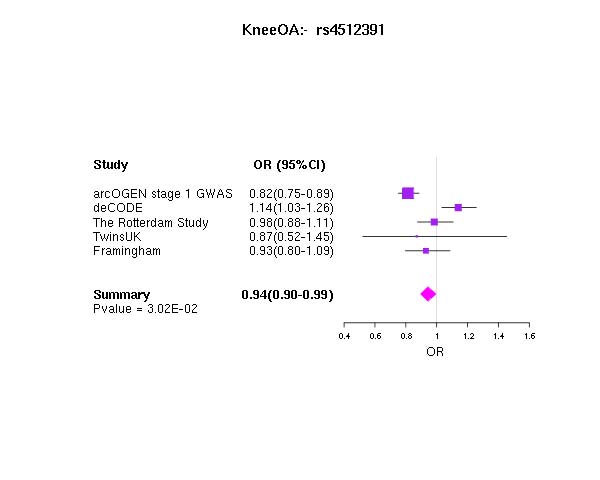
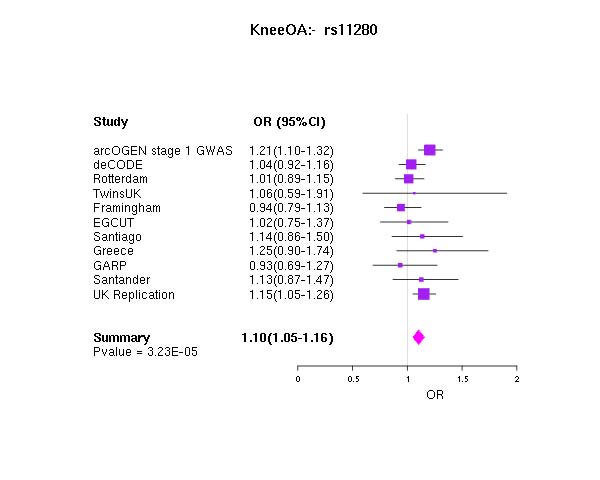
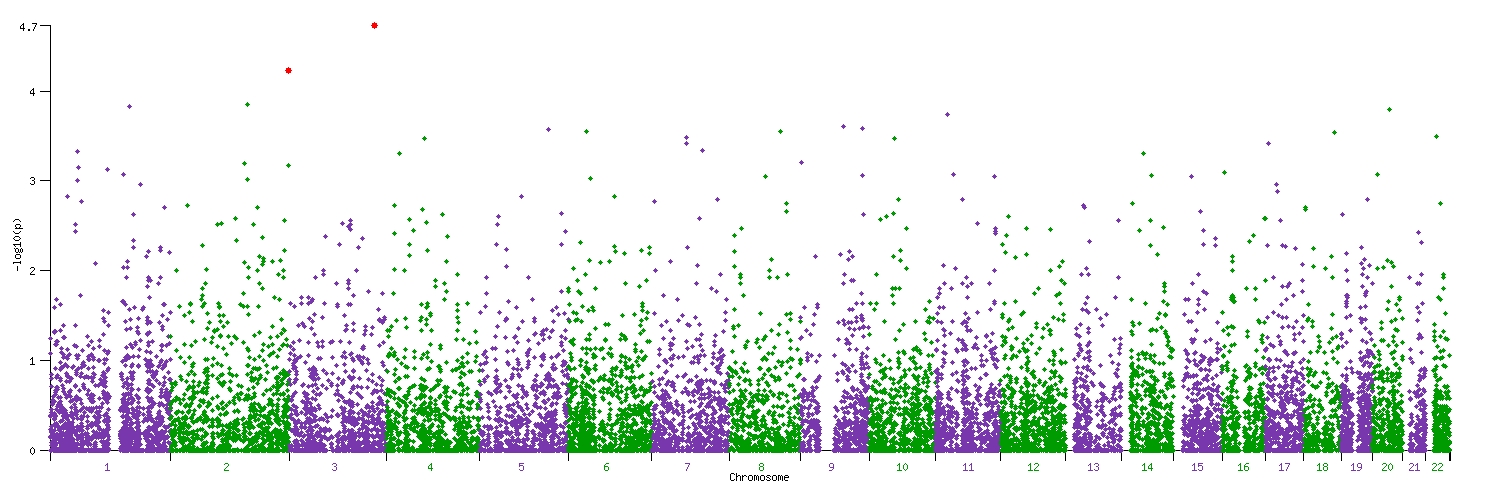
The strongest statistical evidence for association with osteoarthritis was obtained for rs4512391 on chr8 (OR for allele C 1.17; 95% CI 1.10 to 1.25; p=1.8×10−6) approximately 66 kb upstream of the TRIB1 gene. For knee osteoarthritis the most significant finding was also observed at rs4512391 (OR for allele C 1.23, 95% CI 1.13 to 1.33; p=1.1×10−6); for hip osteoarthritis, the strongest signal was rs4977469 (OR for allele A 1.30, 95% CI 1.17 to 1.45; p=1.2×10−6), within intron 3 of the predicted FAM154A gene (supplementary figure 1; supplementary figure 4; supplementary table 2, all available online only). Following replication studies in up to 58 917 independent European-ancestry samples, the overall statistical evidence for association was reduced, with the strongest signal for knee and/or hip osteoarthritis observed at rs2277831 on chr22 (OR for allele G 1.07, 95% CI 1.1.04 to 1.11; combined p=2.3×10−5), within intron 32 of the MICAL3 gene. For knee osteoarthritis the most significant finding post-replication was observed at rs11280 (OR for allele C 1.10, 95% CI 1.05 to 1.16; p=3.2×10−5), within C6orf130; for hip osteoarthritis, the strongest signal was at rs2615977 (OR for allele A 1.10, 95% CI 1.05 to 1.15; p=1.1×10−5) within intron 31 of the COL11A1 gene (supplementary figure 1; supplementary figure 4; supplementary table 2, all available online only). Out of the 65 independent signals (composed of 89 SNP) with p<10−4 in all the osteoarthritis analysis, 17 are present with p<10−4 in the hip osteoarthritis analysis and nine are present with p<10−4 in the knee osteoarthritis analysis, but none reach p<10−4 in both.
Additional analyses, including testing for pairwise interactions, examining overlap between genome-wide association scan and linkage scan signals, association with body mass index, deviation from the additive model, genome-wide HapMap-based imputation, chromosome X association and genome-wide low frequency/rare variant analysis did not identify any additional osteoarthritis signals (supplementary methods; supplementary results, available online only).
The arcOGEN stage 1 GWAS is well powered to detect association with common variants of modest effect at the genome-wide significance level (eg, 90% power to detect an allelic OR of 1.25 at a SNP with frequency 0.35). However, it is poorly powered to detect effects at the established osteoarthritis variants (8% and 4% power for GDF5 and 7q22, respectively), as the index SNP (rs143383, risk allele frequency 0.67; rs3815148, risk allele frequency 0.23) both have small effect sizes (OR ∼1.15).2 3 Retaining the arcOGEN case–control ratio of 1.54, 7774 cases would be required to achieve 80% power to detect an allelic OR of 1.15 at a SNP with risk allele frequency 0.67, and 9084 cases would be required to achieve the same power to detect the same effect size at a SNP with risk allele frequency 0.23.
To evaluate the robustness of association signals, we performed sensitivity analyses using different control sets. We first used ‘supercontrols’ (hip and knee osteoarthritis-free individuals) and found high correlation between association effect estimates (r=0.88) and 93% concordance in the direction of effects (supplementary results, available online only). The two established osteoarthritis loci demonstrated stronger evidence for association compared with the main analysis, even though the ‘supercontrol’ sample size was smaller. We also used a subset of the population-based controls, genotyped on a different platform, to assess robustness to the typing method. We observed 100% concordance in the direction of effect and high correlation between estimates of the OR (r=0.94) (supplementary results, available online only). Therefore, the choice of controls may have affected association strength but not the direction of effect.
The genetic architecture of osteoarthritis is likely to be polygenic with multiple variants along the spectrum of allele frequencies contributing modest and small effects. Our polygene analyses support a model of osteoarthritis in which there is a substantial genetic component comprising multiple contributing variants with small effect sizes (figure 1). SNP with p values as high as 0.25 appear to contribute to the genetic component of osteoarthritis (empirical p=3×10−5, based on ∼85 million permutations), with the bulk of the contribution seen in the 0–0.1 p value range (empirical p=3.5×10−5, ∼101 million permutations). Evaluation of discreet p value bins corroborates this observation and supports a role for SNP with p values up to 0.25 (0.10>p>0.15 bin empirical p=0.06; 0.15>p>0.20 bin p=0.0072; 0.20>p>0.25 bin p=0.022), but with the major contribution coming from SNP with p<0.10 (0>p>0.05 bin p=3×10−5; 0.05>p>0.10 bin p=0.0092). The estimated proportion of variance in disease state explained by the osteoarthritis score alleles is 3.05% (p=3.3×10−4 based on nine million permutations). This is in keeping with findings in other complex common diseases.7

{kind=link}
Summary of polygene analysis results evaluating the genetic architecture of osteoarthritis. Lines in red (real data) and blue (permuted data) show mean±1 SD of the Nagelkerke's pseudo r2 statistic, which indicates the proportion of case–control status accounted for by score alleles in the single-nucleotide polymorphism (SNP) sets. The blue line represents expectation under the null hypothesis of no genetic component to the disease. The red line represents stage 1 genome-wide association study data and shows that SNP at the tail of the p value distribution (up to p<0.25, but primarily p<0.10) have a significantly higher case–control discriminatory capacity.
Discussion
Our stage 1 arcOGEN genome-wide association scan analysis results are in agreement with other large-scale genetic studies,3 4 and clearly indicate that common SNP with large effect sizes are not likely to underpin the aetiology of osteoarthritis. The only two established osteoarthritis loci to date have small OR and our polygene analyses on common SNP suggest that the genetic architecture of osteoarthritis is likely to consist of numerous signals of similar magnitude. The significant increase in sample size with stage 2 of the arcOGEN genome-wide association scan (∼2.4 times as many cases) will increase the power to detect osteoarthritis associations. In addition, large-scale international meta-analysis efforts are underway and will ensure maximal GWAS sample size. A further important parameter in enhancing power and increasing the chances of success involves the improved definition of phenotype. One of the reasons why replication of findings has been difficult to achieve in osteoarthritis could be the inherent heterogeneity of the different osteoarthritis diagnostic and study inclusion criteria used. The field is currently active in evaluating phenotype definition differences and their effects on study power.4
The Wellcome Trust Case Control Consortium first demonstrated the utility of population-based, rather than disease-free, controls in GWAS.8 Our ‘supercontrol’-based sensitivity analyses suggest that for a highly prevalent and heterogeneous disorder such as osteoarthritis, with multiple smaller effects contributing to overall susceptibility, the brute force approach of maximising sample size to balance misclassification in controls is not as successful as it has been for other common diseases, in which ‘low-hanging fruit’ discoveries (of loci with substantial effects) were robust to subtle allele frequency fluctuations.
Osteoarthritis is a heterogeneous disease characterised by variable clinical features with conceivably different genetic aetiologies.9 Although the allele score was associated with osteoarthritis, the proportion of disease variance explained cannot be highly accurately quantified, primarily because of signal attenuation (risk alleles are unlikely to be at the actual causal locus) and sampling variation. The genetic architecture of osteoarthritis is emerging as complex. Large-scale sample sizes and well-defined phenotypes will be required to gain a better understanding of this genetic component, possibly leading to optimising treatment, developing efficacious disease-modifying interventions, improving prognosis and tailoring intervention to the individual.
Supplementary materials
Web Only Data
Files in this Data Supplement:
- View acknowledgements
- View methods
- View table 1
- View table 2
- View table 3
- View table 4
- View table 5
- View table 6
- View table 7
- View table 8
- View table 9
- View table 10
- View table 11
- View table 12
- View figure 2
- View figure 1 page 1
- View figure 1 page 2
- View figure 1 page 3
- View figure 1 page 4
- View figure 1 page 5
- View figure 1 page 6
- View figure 3
- View figure 1 page 1
- View figure 4 page 2
- View figure 4 page 3
- View figure 4 page 4
- View figure 4 page 5
- View figure 4 page 6
- View figure 5
- View figure 6
- View figure 7
- View figure 8
- View figure 9
- View figure 10
- View figure 11
- View figure 12
- View figure 13
- View figure 14
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Footnotes
KP and LS contributed equally. E Zeggini and J Loughlin contributed equally to the study.
-
Funding This study was financially supported by Arthritis Research UK.
-
Competing interests None.
-
Ethics approval This study was conducted with the approval of the Oxfordshire Research Ethics Committee C reference 07/H0606/150.
-
Provenance and peer review Not commissioned; externally peer reviewed