Article Text

Statistics from Altmetric.com
Glucocorticoids are highly effective drugs that are used very widely to inhibit inflammation and to modulate the immune system.1–3 Their great importance in the treatment of various rheumatic diseases is undisputed.4–7 However, although their introduction into clinical medicine was more than 70 years ago, we have understood only a fraction of their mechanisms of action. This is mainly due to their highly pleiotropic effects.8 9 A therapeutically used monoclonal antibody against tumour necrosis factor alpha (TNFalpha) neutralises this molecule, and that is it. In contrast, glucocorticoids as hormones regulate an estimated 20% of the entire genome.10 11 The synthesis of cytokines, chemokines, adhesion molecules, receptors and many enzymes, mediators and other proteins is either upregulated or downregulated. In addition, various mechanisms of their action exist whereby we distinguish between genomic and non-genomic effects. These underlying mechanisms of action are known in principle and discussed in detail elsewhere,3 12–14 so the current knowledge is only summarised here in the form of table 1. However, our understanding of glucocorticoid-mediated immunoregulation still has substantial gaps, not only but especially regarding effects of glucocorticoids in specific cell types and their key cellular targets in particular disease states, and the actions these hormones broadly induce in cells and tissues versus those that are unique to the immune system.9 15
Molecular mechanisms of GC actions3 12–14
Glucocorticoid effects are highly dependent on cell type
Given this background, research in the field of glucocorticoids—sometimes justifiably referred to as ‘old friends’1—continues to be very active. If one enters the search term “glucocorticoids” in PubMed, you will get >230 000 hits. By comparison, the search term “TNF alpha inhibitors” gets only about 56 000 hits. It may be objected that research on glucocorticoids has been going on much longer than that on TNFalpha inhibitors. However, even if you only look at the year 2018, for example, you will find 7900 hits versus 2300 hits. Therefore, it is not surprising that we keep learning unexpected news. For example, the Journal of Experimental Medicine published in 2019 an outstanding paper describing surprising and for rheumatologists and clinical immunologists relevant news in research into the effects of glucocorticoids. Franco et al have investigated the transcriptional effects of glucocorticoids at the level of signalling pathways for nine primary human cell types obtained from healthy donors.11 The cells studied included for example, B cells, CD4+ T cells, monocytes and neutrophils. The authors report that glucocorticoid effects are highly dependent on cell type with regard to the regulation of genes and signalling pathways. They found methylprednisolone to induce cell specific differences in the expression of more than 9000 unique genes or ∼17% of the human transcriptome. I agree with the authors’ interpretation that these results lead to a fundamentally new mechanistic understanding of the effects of glucocorticoids. It is clearly not a one-fits-all concept, but rather that these drugs trigger multifactorial, cell-specific effects. This finding has the potential to develop more selective, cell-specific immunoregulatory therapies.
11b-hydroxysteroid dehydrogenases regulate glucocorticoid effects
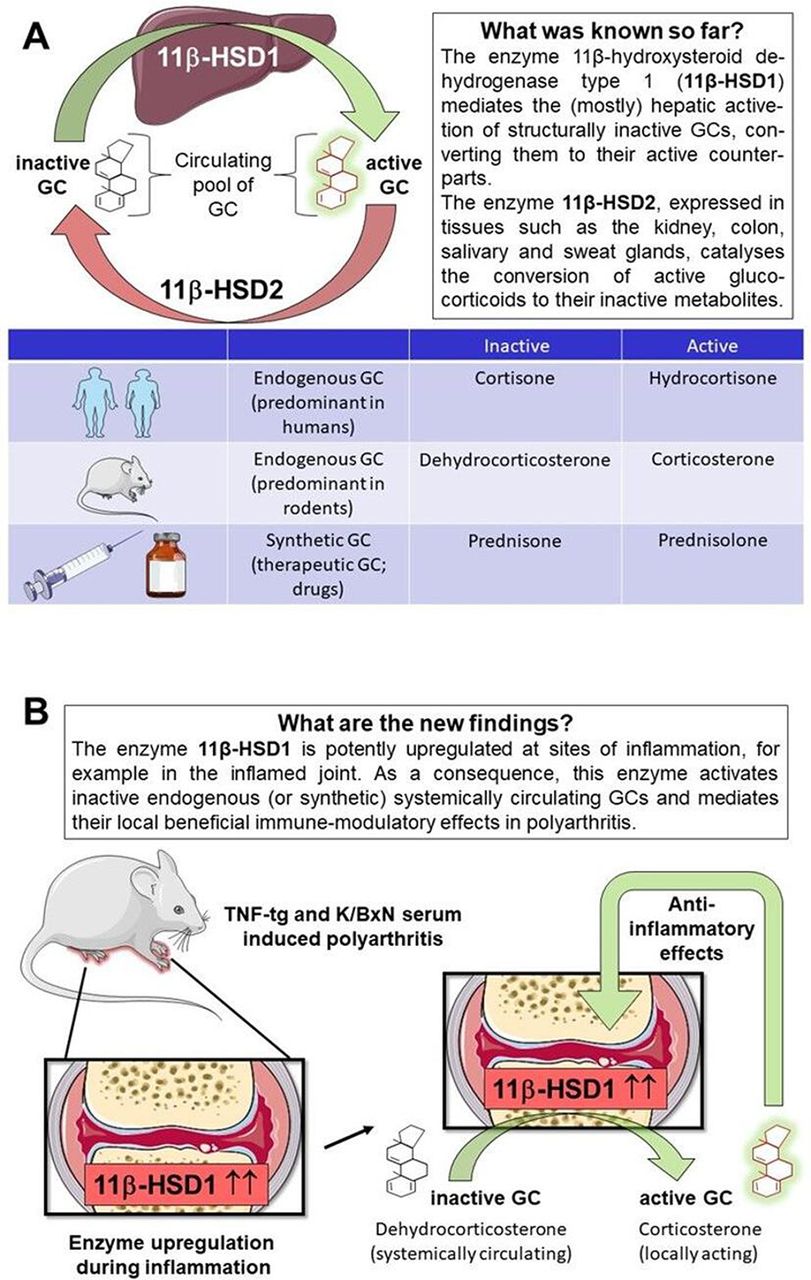
Another example of unexpected significant new findings is published in Annals of Rheumatic Diseases. Fenton et al 16 have dealt with a scientifically rather neglected area in glucocorticoid research, the pre-receptor metabolism. As a background, the type 1 and type 2 (11β-HSD1 and 2), act as key regulators by changing the balance between active and inactive glucocorticoids.17 It is well known that in this way they have a major influence on the expression of glucocorticoid effects in the target cells. Endogenous (ie, physiologically produced) and exogenous (ie, therapeutically given) glucocorticoids circulate in both their active and inactive forms. The (mostly) hepatic 11βHSD1 facilitates the systemic regeneration of biologically active glucocorticoids (cortisol/hydrocortisone, corticosterone, prednisolone) from their inactive forms (cortisone, 11-dehydrocorticosterone, prednisone) by its oxidoreductase (11β reductase) action. Through 11-β dehydrogenation, this enzyme can also facilitate the reverse reaction to some small extent, but it is mainly the (renal) 11βHSD2, which unidirectionally inactivates glucocorticoids (figure 1A).13 18

{kind=link}
The functions of 11β-HSD enzymes in the glucocorticoid (GC) metabolism. (A) GC circulate in both their active and inactive forms. The shuttling between these forms is mediated by the actions of the 11β-HSD enzymes. While mainly the liver 11βHSD1 mediates activation, systemic inactivation is catalysed mainly by renal 11βHSD2. (B) Fenton et al show additionally in murine arthritis models that the peripheral upregulation of 11β-HSD1 in the inflamed joint leads to local reactivation of inactive GC molecules, which induces anti-inflammatory effects.
Fenton et al focused their research not on this systemic, but on the peripheral, intracellular glucocorticoid metabolism in target cells. Both active and inactive glucocorticoid molecules are lipophilic substances that can easily penetrate the cell membrane and thus enter the cytosol of targets such as primary and secondary immune cells, fibroblasts and osteoblasts. However, before they can bind to the cytosolic glucocorticoid receptor alpha to trigger genomic effects (table 1), also in the target cell the activation status is determined by intracellular metabolism via the 11βHSD enzymes. As in the liver, the nicotinamide adenine dinucleotide phosphate (NADPH)-dependent enzyme 11βHSD1 causes local glucocorticoid activation by reduction. In contrast, the 11βHSD2 enzyme catalyses a rapid inactivation by oxidation through the reverse reaction. In order to investigate the contribution of the local 11β-HSD1 enzyme to the local anti-inflammatory properties of glucocorticoids, the authors conducted carefully designed and performed experiments. The primary aim was to measure in murine polyarthritis models the anti-inflammatory properties of orally administered corticosterone in mice with global, myeloid and mesenchymal targeted transgenic deletion of 11β-HSD1. They show that the global deletion of 11β-HSD1 resulted in glucocorticoid treatment being ineffective, proven by findings of persistent synovitis, joint destruction and inflammatory leucocyte infiltration. This was partially reproduced with myeloid 11β-HSD1 deletion (targeted towards neutrophils, macrophages and granulocytes), but not with mesenchymal 11β-HSD1 deletion (targeting primary fibroblasts and osteoblasts). It was also found that paracrine GC signalling between cell populations can overcome targeted deletion of 11β-HSD1. Taken all observations together, the authors conclude that glucocorticoid molecules, which have undergone systemic inactivation, are peripherally, that is, locally in the inflammation area, reactivated by the enzyme 11beta-HSD1 to induce profound anti-inflammatory effects (figure 1B).
This study is certainly of great importance. It reveals a completely new, previously unknown component of the anti-inflammatory effect of glucocorticoids, namely and concisely, that systemically inactivated glucocorticoid molecules are peripherally reactivated by the enzyme 11beta-HSD1 to induce profound anti-inflammatory effects. It should be critically noted, however, that this study has some limitations. First, it must be stressed that results obtained in animal models cannot always be replicated in humans.19 20 Confirmation of the significance of the observations made for clinical medicine is therefore still pending. Second, it is not yet clear whether the identified mechanisms are also applicable to the local activation of therapeutically applied glucocorticoids such as prednisone, prednisolone or methylprednisolone. Third, a more comprehensive scientific picture should be created by including so far previously unconsidered leucocyte subpopulations such as T cells into considerations. Finally, the question remains whether the observations made can be translated into new therapeutic approaches.
It is to be hoped that a deepening of the knowledge gained with regard to glucocorticoid treatment and the proof of its relevance to clinical medicine will in future lead to a further improvement in the therapeutic options for treating patients with rheumatoid arthritis, and other rheumatic and inflammatory diseases, ultimately leading to a better benefit-risk ratio.21 22
References
Footnotes
Handling editor Josef S Smolen
Funding The author has not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Patient consent for publication Not required.
Provenance and peer review Commissioned; externally peer reviewed.